Translate this page into:
Dystrophin-associated muscular dystrophies: Learning from genetics to guide therapeutics
Corresponding Author:
Naif A.M. Almontashiri
Center for Genetics and Inherited Diseases, Taibah University, P.O. Box 42318, Almadinah Almunwwarah
Saudi Arabia
nmontashri@taibahu.edu.sa
| How to cite this article: Almontashiri NA. Dystrophin-associated muscular dystrophies: Learning from genetics to guide therapeutics. J Musculoskelet Surg Res 2019;3:241-244 |
Abstract
Dystrophin-associated muscular dystrophies represent an X-linked group of disorders, with spectrum of clinical phenotypes caused by mutations in the Duchenne muscular dystrophy (DMD) gene. Based on the genotype and predicted impact of the mutations on the function and expression of dystrophin, the clinical phenotype ranges from severe-to-mild muscular diseases, namely DMD, Becker muscular dystrophy, and dilated cardiomyopathy. Diagnosis, prediction of severity, and management of dystrophinopathies are largely based on the type of mutations and their impact on the dystrophin expression or function. In this review, we highlight the clinical and molecular etiologies and spectrum of the dystrophin-associated muscular dystrophies. We provide an overview on the molecular testing approach for the molecular diagnosis of cases with suspicion of or clinical diagnosis of dystrophinopathies. Finally, we provide an updated summary of the current therapeutic advances using gene therapy and editing technologies to treat or mitigate dystrophin-associated muscular dystrophies.Introduction
The mild and severe forms of dystrophin-associated muscular dystrophies include Becker muscular dystrophy (BMD), dilated cardiomyopathy (DCM), and Duchenne muscular dystrophy (DMD). The phenotype can be as mild as muscle cramps accompanied with creatine phosphokinase elevation and myoglobinuria in asymptomatic carrier to progressive skeletal and heart muscle dystrophies in those affected with DMD and BMD.[1] Although this is an X-linked recessive group of disorders where males are predominantly and can be severely affected, carrier females who are usually asymptomatic can be as severely affected as males. This might be partly explained by the skewed chromosome X-inactivation.[2],[3]
DMD is the severe, lethal, and early-onset dystrophinopathy. It manifests with delay in motor development and progressive proximal muscle weakness, rendering those affected wheelchair-bound by age 12. Mild-to-severe mental retardation has been reported in ~20% of the patients.[4] Few survive DMD with the respiratory and DCM-related complications being the most common causes of death in those patients. On the milder side, BMD manifests with a later onset of muscle weakness with DCM-related heart failure being the most common cause of death in those patients. Some male patients with mutations in the DMD gene present with isolated DCM leading to congestive heart failure with carrier females being at risk of developing DCM, making molecular testing and the following clinical surveillance very useful.[5]
Molecular Mechanism and Testing of Dystrophinopathies
Dystrophin-associated muscular dystrophies are caused by mutations in the X-linked DMD gene (Gene MIM number: 300, 377) leading to absent or dysfunctional dystrophin. Dystrophin is mainly expressed in skeletal, cardiac muscles, and; to lesser extent, some brain cells. In muscle cells, the dystrophin-protein complex anchors and connects the cytoskeleton with the extracellular matrix to provide strength to muscle fibers and protection against damage induced during the contraction-relaxation of muscles.[6] In addition, dystrophin plays an important role in the proper structure and function of synapses and acts as scaffolding for signaling molecules required for neurotransmitters release.[7]
Molecular diagnosis of dystrophinopathies can be confirmed by detecting hemizygous or heterozygous pathogenic or likely pathogenic variants in the DMD gene in males and females, respectively. The most common type of variants is large deletions and duplications, accounting for 60% of the cases, whereas the rest of the cases are accounted for by small frameshift and missense variants.[8] It is noteworthy that the DMD gene is the largest human gene of about 2.2-ml base pairs in size, making the mutation spectrum wide and hard to test using one single diagnostic assay.[9] Therefore, the diagnostic molecular assay to be used varies based on the type of mutation to be detected. The sensitive quantitative assays, quantitative polymerase chain reaction, multiplex ligation-dependent probe amplification, and gene-targeted microarray are widely used and recommended to detect large- and exon-level deletions and duplications. The gene-targeted deletion/duplication assays can detect the causal variants in ~65%–80% of dystrophinopathies.[5] If negative, gene sequencing or multigene panel assays using next-generation sequencing, for example, are to be considered for detecting small frameshift and point variants, whereas assays such as Sanger sequencing can be utilized to detect known familial variants (targeted testing).[5],[10] Gene sequencing assays can detect the causal variants in ~20%–35% of dystrophinopathies.[5]
According to the guidelines of the American College of Medical Genetics and Genomics,[11] variants detected by molecular testing can be classified into three major categories: (1) benign or likely benign variants: this category of variants is considered “negative genetic testing” and therefore requires additional molecular testing (to find the disease-causing mutations that are missed by the current assay), clinical reevaluation, and testing (skeletal muscle biopsy for immunohistochemistry and Western blot studies of dystrophin) or considering other types of muscular dystrophies with different causal genes; (2) pathogenic and likely pathogenic variants: this category of variants is considered “positive genetic testing” and sufficient to establish a molecular diagnosis; however, clinical correlation and segregation studies are strongly recommended; and (3) variants of unknown or uncertain significance: this category of variants is considered “inconclusive genetic testing,” and therefore, require additional clinical (see Category 1), biochemical, and segregation studies to exclude or prove pathogenicity [Figure - 1].
![[Figure - 1]](#fig_SaudiOrthopJ_2019_3_3_241_263825_f1.jpg){kind=link}
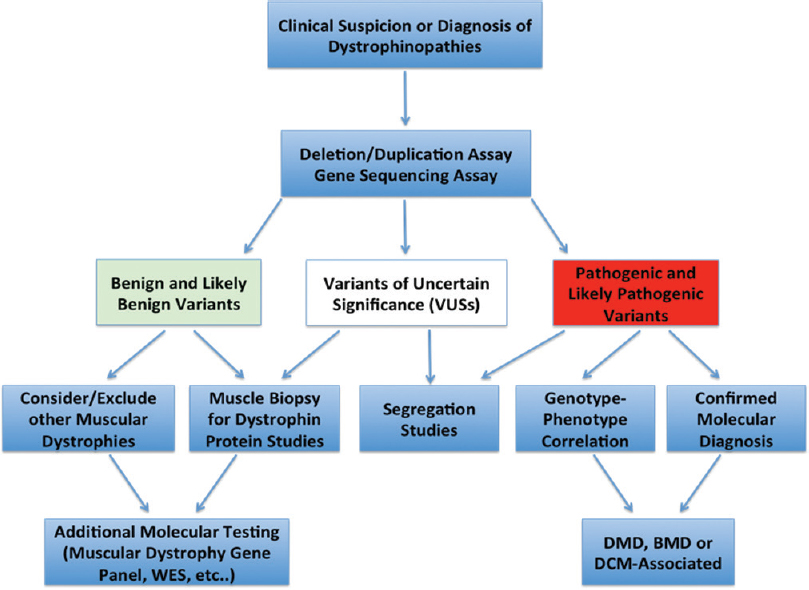 |
| Figure 1: Molecular diagnostic algorithm for dystrophinopathies. WES: Whole-exome sequencing, DMD: Duchenne muscular dystrophy, BMD: Becker muscular dystrophy, DCM: Dilated cardiomyopathy |
There are several reputable and easy-to-use mutations databases that can be used by scientists and clinicians for curating, classifying, and reading-frame checking, when interpreting a given variant in the DMD gene [Table - 1].
![[Table - 1]](#tbl_SaudiOrthopJ_2019_3_3_241_263825_t2.jpg){kind=link}

Genotype–phenotype Correlation
The phenotypic spectrum of the DMD variants follows the “open reading frame” rule, except for about < 10% of the cases.[12] Variants that will alter the reading frame by the insertion, duplication, and deletion (frameshift variants) of nucleotides will shift the reading frame and introduce a premature termination codon (PTC), which will subject the mRNA to a selective degradation through the nonsense-mediated decay (NMD) pathway, most likely leading to absent or sometime truncated protein. The frameshift variants in the DMD gene are well correlated with and can be used to predict and differentiate Duchenne from BMD and also excluding the need for muscle biopsy in most of the cases.[13] In contrast, point (nontruncating) and splicing variants that do not alter the reading frame by insertion, duplication, and deletion (in-frame mutations) of three (or multiples of three) nucleotides will result in shorter or longer protein with reduced function and; yet enough to correlate well with the milder BMD.[13]
Interestingly, in addition to variants in exon 1 of the DMD gene, variants in the promoter, required for muscle-specific expression of dystrophin, are generally associated with DCM without affecting the skeletal muscle.[14],[15],[16],[17] This is because of the presence of actively expressed alternative, nonmuscle (central nervous system) promoters in the skeletal muscle (not cardiac muscle), and produces dystrophin enough for the skeletal muscle to be not affected by the impairment of the muscle-specific promoter. However, other variants in other exons have been previously reported in patients with isolated DCM.[5],[18],[19]
Management
The management of dystrophin-associated muscular dystrophies is mainly focused on the manifestations of the disease such as DCM, congestive heart failure, scoliosis, and muscle strength. The successful management also includes surveillance and prevention of secondary respiratory and cardiac and musculoskeletal complications. Given the lethality and significant health burden associated with dystrophinopathies, genetic counseling is imperative for families with a history of dystrophin-associated muscular dystrophies to help them support the living affected members and avoid recurrences by utilizing the available options, such as preimplantation genetic diagnosis and prenatal genetic testing.
Therapies
Research and clinical studies are currently underway to find novel ways to repair or restore the dystrophin gene and protein, hoping to cure or mitigate BMD and DMD. Several approaches are taken to tackle different types of DMD variants. Basically, for the dystrophin to be restored or corrected, adeno-associated viruses (and other molecular delivery vehicles, such as exosomes) are used to deliver the dystrophin gene or dystrophin-editing tools to correct or skip mutations.[20]
Several investigative therapies using exon skipping with antisense oligonucleotides or read through of PTCs to bypass or suppress NMD, correct the DMD reading frame, and restore dystrophin expression. Many of these investigative therapies have passed the preclinical studies and showed muscle benefits and improvements in clinical studies.[5],[9] However, the need for the readministration of doses and lack of satisfying motor clinical benefits are limitations of such approaches. To address these limitations, gene repair using powerful gene-editing tools such as clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (CRISPR-Cas9) technology is needed to specifically and permanently target and correct the DMD variants inside the relevant cells, where dystrophin is mostly needed. Indeed, promising preclinical studies on mice and dogs models of DMD have been conducted and showed very encouraging curative results.[21],[22],[23] For example, in four dogs, CRISPR gene editing was successful in restoring ~ 90% and 92% of normal dystrophin levels in skeletal and cardiac muscles, respectively. These levels were correlated with improved muscle histology and motor function.[24],[25]
The progress in DMD gene editing has paved the way for upcoming clinical trials to use this technology to cure dystrophin-associated muscular dystrophies in human. The large size of the DMD gene and safety and specificity of the gene editing and delivery tools are examples of obstacles ahead of the clinical trials that need to be addressed at the biological and technical levels.
Conclusion
The diagnosis, management, and treatment of dystrophinopathies should be coupled and correlated with genetic testing and findings. This approach, with the reducing cost of sequencing and genetic testing, will save cost, time, and provide proper management for timely intervention and counseling. Genetic testing for dystrophinopathies is not just a molecular confirmation but also a clinical need for precision medicine.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Authors' contribution
NAM prepared the drafted and final version of the review article. NAM has reviewed and approved the final draft and, therefore, responsible for the content and similarity index of the manuscript. The author has critically reviewed and approved the final draft and is responsible for the content and similarity index of the manuscript.
| 1. | Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731-40. [Google Scholar] |
| 2. | Giliberto F, Radic CP, Luce L, Ferreiro V, de Brasi C, Szijan I, et al. Symptomatic female carriers of duchenne muscular dystrophy (DMD): Genetic and clinical characterization. J Neurol Sci 2014;336:36-41. [Google Scholar] |
| 3. | Viggiano E, Picillo E, Ergoli M, Cirillo A, Del Gaudio S, Politano L, et al. Skewed X-chromosome inactivation plays a crucial role in the onset of symptoms in carriers of becker muscular dystrophy. J Gene Med 2017;19. Doi: doi.org/10.1002/jgm.2952. [Google Scholar] |
| 4. | Bushby KM, Appleton R, Anderson LV, Welch JL, Kelly P, Gardner-Medwin D, et al. Deletion status and intellectual impairment in duchenne muscular dystrophy. Dev Med Child Neurol 1995;37:260-9. [Google Scholar] |
| 5. | Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA): GeneReviews Database; 1993. [Google Scholar] |
| 6. | Biggar WD, Klamut HJ, Demacio PC, Stevens DJ, Ray PN. Duchenne muscular dystrophy: Current knowledge, treatment, and future prospects. Clin Orthop Relat Res 2002;(401):88-106. [Google Scholar] |
| 7. | Anderson JL, Head SI, Rae C, Morley JW. Brain function in duchenne muscular dystrophy. Brain 2002;125:4-13. [Google Scholar] |
| 8. | Hoffman EP, Dressman D. Molecular pathophysiology and targeted therapeutics for muscular dystrophy. Trends Pharmacol Sci 2001;22:465-70. [Google Scholar] |
| 9. | Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: Pathogenesis and opportunities for treatment. EMBO Rep 2004;5:872-6. [Google Scholar] |
| 10. | Abbs S, Tuffery-Giraud S, Bakker E, Ferlini A, Sejersen T, Mueller CR, et al. Best practice guidelines on molecular diagnostics in duchenne/Becker muscular dystrophies. Neuromuscul Disord 2010;20:422-7. [Google Scholar] |
| 11. | Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405-24. [Google Scholar] |
| 12. | Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the leiden duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006;34:135-44. [Google Scholar] |
| 13. | Deburgrave N, Daoud F, Llense S, Barbot JC, Récan D, Peccate C, et al. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum Mutat 2007;28:183-95. [Google Scholar] |
| 14. | Bastianutto C, Bestard JA, Lahnakoski K, Broere D, De Visser M, Zaccolo M, et al. Dystrophin muscle enhancer 1 is implicated in the activation of non-muscle isoforms in the skeletal muscle of patients with X-linked dilated cardiomyopathy. Hum Mol Genet 2001;10:2627-35. [Google Scholar] |
| 15. | De Repentigny Y, Marshall P, Worton RG, Kothary R. The mouse dystrophin muscle enhancer-1 imparts skeletal muscle, but not cardiac muscle, expression onto the dystrophin purkinje promoter in transgenic mice. Hum Mol Genet 2004;13:2853-62. [Google Scholar] |
| 16. | Milasin J, Muntoni F, Severini GM, Bartoloni L, Vatta M, Krajinovic M, et al. Apoint mutation in the 5' splice site of the dystrophin gene first intron responsible for X-linked dilated cardiomyopathy. Hum Mol Genet 1996;5:73-9. [Google Scholar] |
| 17. | Neri M, Valli E, Alfano G, Bovolenta M, Spitali P, Rapezzi C, et al. The absence of dystrophin brain isoform expression in healthy human heart ventricles explains the pathogenesis of 5' X-linked dilated cardiomyopathy. BMC Med Genet 2012;13:20. [Google Scholar] |
| 18. | Chamberlain RC, Smith EC, Campbell MJ. Novel rod domain duplication in dystrophin resulting in X-linked dilated cardiomyopathy. Pediatr Neurol 2015;53:439-41. [Google Scholar] |
| 19. | Shimizu M, Ino H, Yasuda T, Fujino N, Uchiyama K, Mabuchi T, et al. Gene mutations in adult Japanese patients with dilated cardiomyopathy. Circ J 2005;69:150-3. [Google Scholar] |
| 20. | Levin AA. Treating disease at the RNA level with oligonucleotides. N Engl J Med 2019;380:57-70. [Google Scholar] |
| 21. | Koch L. Genetic engineering:In vivo genome editing – Growing in strength. Nat Rev Genet 2016;17:124. [Google Scholar] |
| 22. | Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016;351:400-3. [Google Scholar] |
| 23. | Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science 2016;351:403-7. [Google Scholar] |
| 24. | Amoasii L, Hildyard JC, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, et al. Gene editing restores dystrophin expression in a canine model of duchenne muscular dystrophy. Science 2018;362:86-91. [Google Scholar] |
| 25. | Stower H. Gene editing for duchenne muscular dystrophy. Nat Med 2018;24:1491. [Google Scholar] |
Fulltext Views
2,918
PDF downloads
1,580




