Translate this page into:
Isolated congenital vertical talus: Genetics and genomics
2 Center for Genetics and Inherited Diseases, Taibah University, Almadinah Almunnawarah, Saudi Arabia
Corresponding Author:
Sulman Basit
Center for Genetics and Inherited Diseases, Taibah University, Almadinah Almunnawarah
Saudi Arabia
sbasit.phd@gmail.com
| How to cite this article: Khan YN, Basit S. Isolated congenital vertical talus: Genetics and genomics. J Musculoskelet Surg Res 2020;4:66-71 |
Abstract
Congenital vertical talus (CVT) is a distinct orthopedic condition where the bone structure and number are normal in the affected foot, but the orientation of the bones is not correct. The abnormal orientation of the bones in the affected foot is believed to be due to muscle imbalance. There are a shortening and dorsal displacement of the peroneal tendons and tibialis posterior tendon, resulting in the clinical appearance of a severe rigid flatfoot. The underlying etiology of the CVT is unknown, and limited studies have been performed to decipher the genetics of CVT. The purpose of this review is to highlight the key research articles within the CVT genetics and genomics fields that were published previously. Herein, we reviewed the current literature and discussed the genetic studies carried out in families and patients with an isolated form of CVT. It is believed that CVT segregates in an autosomal dominant fashion. Most of the studies used a candidate gene approach to identify CVT causative variants. Variants in growth differentiation factor 5, HOXD10, teashirt zinc finger homeobox 1, and skeletal muscle contractile genes have been associated with CVT. An unbiased and hypothesis-free approach of whole-exome sequencing is much needed to unwire the genetic network underlying distal hind limb development and to improve our understanding of the gene regulatory mechanism in this musculoskeletal disorder. Moreover, this review focuses on highlighting the importance of the identification of the genetics of CVT and its implications in early clinical diagnosis and management of patients.
Introduction
A large number of musculoskeletal abnormalities exist, which affect the morphology and growth of one or group of bones, muscles, and connective tissues in the skull, trunk, and limbs. These abnormalities ultimately affect the integrity of body position, weight-bearing property, and locomotion. Musculoskeletal abnormalities and disorders of limbs form the largest group of congenital defects.[1] Congenital limb deformity is frequent with variable clinical presentation and can present as an isolated disorder (nonsyndromic) or a part of a syndrome (associated with other extra-musculoskeletal conditions). Congenital vertical talus (CVT) is a rare congenital limb deformity that affects the positioning of the foot.[2] It is characterized by valgus and equinus deformity of the hindfoot, along with the dorsiflexion at the midfoot and abduction of the forefoot.[3] This abduction is caused by a fixed dorsal dislocation of the navicular bone on the head of the talus. Typical CVT is considered as a type I CVT. However, it is termed as CVT type II if the CVT type I includes deformity of the calcaneocuboid joint.[4],[5] The incidence of a CVT is 1 in 10,000, and it affects both males and females in equal proportion.[2],[5] CVT is bilateral in approximately 50% of cases.[2],[6] CVT rarely occurs as a nonsyndromic condition and in most of the cases exists as a part of a syndrome.[7],[8] Syndromic forms of CVT are mostly associated with defects of the central nervous system (arthrogryposis, myelomeningocele, sacral agenesis, and neurofibromatosis), muscle abnormalities (ischiocalcaneal band), acquired deformities (cerebral palsy and spinal muscular atrophy), and certain genetic conditions (Freeman–Sheldon syndrome, Smith–Lemli–Opitz syndrome, Marfan syndrome, Sheldon–Hall syndrome, nail–patella syndrome, Eagle-Barrett syndrome, and split-hand/split-foot malformation).[5],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18]
Clinical Presentation of Congenital Vertical Talus
CVT is also frequently termed as “congenital convex pes valgus” or “rocker-bottom foot” deformity.[19],[20],[21] CVT is a dislocation of the talonavicular joint characterized by vertical positioning of the talus with a rigid dorsal dislocation of the navicular bone, equinus malformation of the calcaneum, abduction defect of the forefoot, and soft-tissue contracture of the hindfoot and midfoot.[21] The Achilles tendon is contracted, and the calcaneum is in equinus.[22] Radiographic evaluation on the lateral view shows the long axis of the talus to be vertical and lying parallel with the longitudinal axis of the tibia.[22] Forced plantar flexion and forced dorsiflexion lateral radiographs are also required for the confirmation of the CVT diagnosis and to differentiate it from the oblique talus. The forced plantar flexion lateral view shows persistent malalignment of the long axis of the talus and the first metatarsal [Figure - 1]. The forced dorsiflexion lateral radiograph exhibits a persistently decreased tibiocalcaneal angle showing fixed hindfoot equinus and shows the persistent malalignment of the long axis of the talus in relation to the navicular bone.[22] A postmortem examination on newborns, as well as findings during surgical corrections, has contributed to our understanding of the pathoanatomy of the CVT.[3],[19],[23],[24] Dorsal and lateral displacements of the navicular bone with respect to the head and neck of the talus were observed. Moreover, the navicular bone becomes hypoplastic and wedge shape due to abnormal articulation with the talus. For a definite diagnosis of a case, it is essential to maintain the foot in extreme plantar flexion to show that the navicular bone is dislocated dorsally on the neck of the talus.[25] Contractures of foot muscles, including tibialis anterior, extensor hallucis brevis, peroneus tertius, peroneus longus, and peroneus brevis, were frequently observed.[22]
![[Figure - 1]](#fig_SaudiOrthopJ_2020_4_2_66_281899_f1.jpg){kind=link}
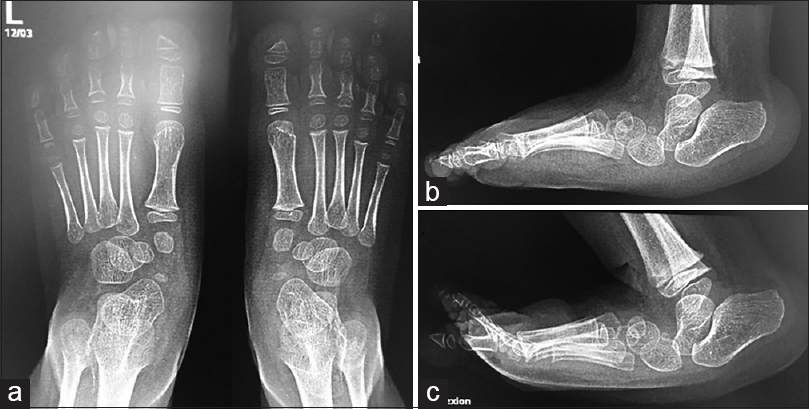 |
| Figure 1: (a) Anteroposterior of 3-year-old girl feet. (b and c) Lateral view demonstrates the malalignment of the first ray with the talus bone. The long axis of the talus passes planter to the metatarsal axis |
An underlying neuromuscular disease must be ruled out in isolated CVT.[7] In order to rule out isolated CVT, any evidence of flexion contractures or ulnar deviation of the fingers, as well as limitation of motion at other joints, must be excluded. Radiographic assessment is enough to establish the diagnosis. CVT has been associated with certain defects of the central nervous system, multiple malformation syndromes, in utero deformations, and some gross chromosomal defects.[4] However, the precise etiology of CVT is still unknown.
Treatment of Congenital Vertical Talus
CVT is treated surgically as well as nonsurgically. The main aim of the treatment is to resume the normal function of the foot by correcting the anatomic relationship among talus, navicular bone, and calcaneum. Most commonly employed nonsurgical treatment methods involve conservative therapies, such as manipulation and serial casting.[26] This method is used to improve the deformity and thus decrease the complexity of the surgical procedure.[25] However, these methods are not always successful in this deformity.[6] The surgical procedure involves correction between the ages of 6 and 12 months. Mostly, a single-stage surgery is used to obtain the necessary correction.[25],[27],[28] The use of consecutive plaster cast treatment (serial cast correction) to slowly reduce the talonavicular joint, followed by minimal surgical interventions, has shown good early results in the treatment of CVT.[26],[29]
Transmission of Congenital Vertical Talus
Isolated forms of CVT have been reported to transmit in families. In most of the familial cases of CVT, an autosomal dominant inheritance has largely been considered as a transmission pattern.[7],[30],[31],[32],[33] Families with asymptomatic parents have also been reported.[32] However, in cases where both parents are asymptomatic, the inheritance pattern can also be considered as an autosomal recessive. Nevertheless, in such cases, carriers were presumed to be nonpenetrant. Definite inheritance pattern could not be determined in familial CVT due to small family size, the rarity of the problem, incomplete penetrance of the phenotype, and lack of thorough clinical evaluation of carriers.
Genetics of Congenital Vertical Talus
The contribution of genetic factors in the etiology of CVT is evident from the fact that 50% of isolated CVT patients have affected first-degree relatives.[30] Several families with more than one affected individual segregating CVT have been reported. Moreover, the association of CVT with well-established genetic syndromes strengthens the hypothesis of genetic factors underlying CVT. The segregation of CVT in families suggests a major role of a single gene variant(s) in an individual family. In this context, defects in signaling pathways and the associated molecular components involved in the development of the skeleton might be the dominant players and an underlying cause of CVT.
Genetic Models Used to Decipher the Genetics of Congenital Vertical Talus
Genetic association, as well as candidate gene variant approaches, has been used to identify the genetic factors underlying CVT. Moreover, a genome-wide linkage analysis study design has also been used to decode the genetics of CVT.
Candidate Gene Sequencing
Cartilage-derived morphogenetic protein-1(growth differentiation factor 5)
Cartilage-derived morphogenetic protein-1 (CDMP1) is also known as growth differentiation factor 5. CDMP1 encodes a ligand for the transforming growth factor-beta (TGF-β) superfamily of proteins. Binding of CDMP1 ligand with the TGF-β receptors leads to the recruitment and activation of a group of transcription factors and thus controls gene expression in various cell types and subsequently plays a role in the development of cartilage, joints, and the growth of neuronal axons.[35],[36],[37],[38] Mutations in CDMP1 cause severe upper- and lower-limb malformations, including Grebe-type acromesomelic dysplasia, Hunter-Thompson-type chondrodysplasia, Du Pan syndrome, and brachydactyly type C.[35],[39],[40],[41],[42],[43],[44],[45],[46] Some reports also showed that CVT is a part of a spectrum of skeletal malformations due to CDMP1 heterozygous mutations.[35],[42] Candidate gene sequencing approach was used in families with CVT, and a heterozygous mutation (c.1312C>T; p.R438C) in CDMP1 was identified in a North American family with isolated CVT.[47] The data have not been replicated, and this is the only report available. The authors of this review have performed in silico analysis of the variant (c.1312C>T) and found that the variant is in the active domain of the CDMP1 and is predicted to be likely disease causing. For instance, it is predicted to be deleterious by sorting intolerant from tolerant (SIFT) (score 0), probably damaging by PolyPhen-2 (score 1), and damaging by MetaLR (score 0.738) tools (unpublished data).
HOXD10
HOXD10 gene encodes a homeobox DNA-binding domain containing protein. The HOXD10 protein is expressed in the limb buds during development and is known to play a role in differentiation and limb development functions by acting as a sequence-specific transcription factor.[48],[49] Mutations in the HOXD10 gene have been associated with Wilms' tumor,[9] esophageal squamous cell carcinoma,[42] and CVT.[33],[34],[43],[44] A missense mutation (c.956T >A; p. Met319Lys) in the homeodomain recognition helix of HOXD10 was detected in all affected individuals of an extended family with isolated CVT.[34] This mutation was previously reported in a family with bilateral CVT and Charcot–Marie–Tooth disease.[50],[51] The authors of this review have performed in silico analysis of the variant (c.956T>A) and found that the variant is pathogenic (ClinVar) and clinically significant. It is expected to be deleterious by SIFT (score 0), possibly damaging by PolyPhen-2 (score 1), and damaging by MetaLR (score 0.9496) tools (unpublished data). However, a significant number of families with isolated CVT are negative for mutations in the coding as well as 5' and 3' untranslated regions of the HOXD10 gene.[52] This shows that HOXD10 mutations are not a common cause of isolated CVT. Therefore, downstream spatiotemporal transcriptional targets of the HOXD10 gene must be characterized and screened in CVT families. HOXD10 target genes expressing in the developing limb may be the excellent candidate genes for CVT.
Copy Number Variation and Congenital Vertical Talus
The deletion of the distal part of the long arm of chromosome 18 (18q deletions) is known to cause the 18q-deletion syndrome. The 18q-deletion syndrome phenotype has been described well and varies greatly among individuals with 18q deletions.[53],[54] Variations in clinical features in patients with distal 18q deletions are due to the difference in the size of the deletion. Various studies have used chromosomal microarray to identify the precise genotype–phenotype correlation in patients with 18q deletions.[53],[54],[55],[56],[57],[58],[59],[60],[61] The region was narrowed to a 5.8 Mb segment (69.1–74.9 Mb) for lower-limb deformities, including CVT.[60] It was further narrowed to 1.70 Mb (72.2–73.9 Mb) containing just five genes (ZNF407, ZADH2, teashirt zinc finger homeobox 1 (TSHZ1), C18orf62, and ZNF516) in patients with bilateral CVT features only [Figure - 2].[62] Further analysis of the deletions using patient data from the DECIPHER database (http://decipher.sanger.ac.uk/) and data published by Feenstra et al. (2007) refined the region to 1.02 Mb (72.9–73.5). This region contains only TSHZ1 and C18orf62 genes [Figure - 2].
![[Figure - 2]](#fig_SaudiOrthopJ_2020_4_2_66_281899_f2.jpg){kind=link}
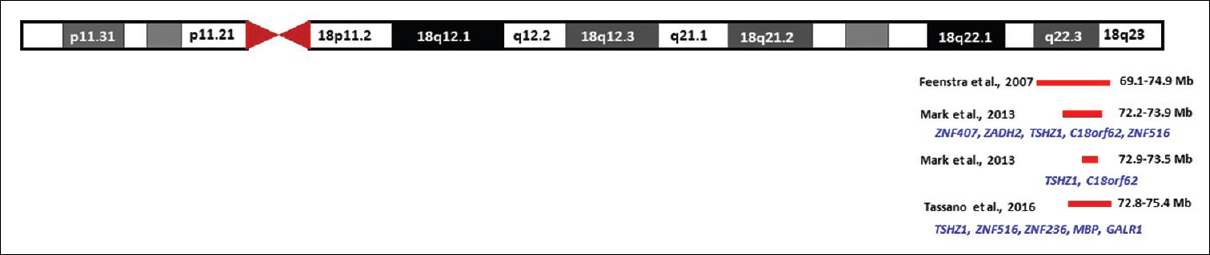 |
| Figure 2: Schematic representation of the congenital vertical talus critical genomic region in 18q deletion |
Teashirt zinc finger homeobox 1 and C18orf62
TSHZ1 encodes a transcriptional factor containing atypical DNA-binding domain.[63],[64] It is predicted to be involved in the developmental processes through transcriptional regulation of target genes.[64] Based on the expression pattern of the TSHZ1 in human tissues and its role in murine skeletal growth and development, the gene TSHZ1 was considered as a likely candidate gene for the bilateral CVT phenotype in 18q-deletion syndrome.[62] A recent study has identified a 2.5 Mb deletion (chr18: 72.8–75.4) in a patient with syndromic features, including bilateral CVT using array comparative genomic hybridization.[65] The region overlaps with the previously identified region and has TSHZ1 as the only common gene [Figure - 2]. Therefore, TSHZ1 is the most plausible candidate gene for CVT in the 18q-deletion syndromes. The second gene in the 1.02 Mb (72.9–73.5) critical region on chromosome 18 is C18orf62 or small integral membrane protein 21 (SMIM21). SMIM21 is an uncharacterized protein-coding gene. Expression data are lacking, and pathogenic variations in this gene have not been associated with any human disease. Moreover, this gene is not present in the smallest common deletions identified by Mark et al. and Tassano et al.[62],[65] Therefore, up till now, the impact of the heterozygous deletion of SMIM21 on the clinical features of CVT is unclear.
Skeletal Muscle Contractile Genes in Congenital Vertical Talus
Contractile genes encode a component of the contractile apparatus of skeletal myofibers. It is known that the muscle biopsy specimens from patients with CVT have abnormalities in skeletal muscles, including the small size of the muscle fiber and predominant abnormal fiber type.[66] Mutations in skeletal muscle contractile genes (MYH3, MYH8, TPM2, TNNI2, and TNNT3) are responsible for distal arthrogryposis (DA).[66],[67],[68],[69],[70] The DA syndrome is a group of abnormalities manifested as nonprogressive congenital contractures mainly involving the distal parts of the limbs.[71],[72],[73] The foot phenotype described in individuals with DA is similar to the foot features in isolated CVT.[71] Therefore, it is proposed that the contractile genes responsible for DA might also be involved in more common distal limb defects, including CVT.[74] This assumption is supported by the fact that variations in skeletal muscle contractile genes influence the risk of clubfoot, another distal limb anomaly.[69] Therefore, skeletal muscle contractile genes must be considered in the etiology of CVT. However, the resequencing of coding exons of three contractile genes (MYH3, TNNT3, and TPM2) failed to identify any pathogenic variant in CVT patients.[74] Failure to identify the CVT-causing variants in contractile genes has been attributed to the low number of samples tested.[74]
Discussion
CVT occurs both in isolated and syndromic forms. Therefore, it is important to rule out the CVT-associated clinical features while assessing individuals with apparently isolated forms of CVT. Extra-musculoskeletal features may also exist with CVT, including neurological malformations. Approximately half of the CVT cases are associated with abnormalities of various systems including muscles, skeleton, and nervous system.[2],[9],[11],[13],[14],[31],[75],[76],[77],[78]
The role of genetic variations in the etiology of CVT is evident from the fact that CVT has the tendency to aggregate and segregate in families. Moreover, the autosomal dominant inheritance model has been widely suggested emphasizing Mendelian segregation and monogenic factor within a family. Furthermore, isolated CVT patients with a first-degree relative have been reported in 50% of the cases.[30] The genetic factors underlying CVT are not fully penetrant, and a variable expression has been observed in multiple families segregating CVT.[7],[34] Although genetic variants are implicated in CVT, the effect of these variants in the pathogenesis of CVT is estimated to be small to moderate in size and may vary from population to population.
Variants in HOXD10 and CDMP1 have been associated with CVT. However, the replication studies are largely missing. The deletion of TSHZ1 gene has also been implicated in isolated CVT, though heterozygous knockout mice failed to recapitulate human phenotype.[64] Several studies failed to identify defects in genes associated with CVT. This emphasizes that CVT is a genetically heterogeneous phenotype, and large-scale studies are required to decipher the gene network underlying CVT phenotype. We suggest whole-exome sequencing in those families where at least 2 individuals with CVT are available. Once an underlying gene CVT is identified, the gene can be screened in isolated cases. Besides, in isolated cases, copy number variation detection using dense SNP array might lead to detect indels underlying CVT.
Ethical approval
The study was approved by the ethical review committee of the college of medicine, Taibah University Almadinah Almunawwarah.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient's parents have given their consent for the images and other clinical information to be reported in the journal. The parents understand that the name and initials will not be published and due efforts will be made to conceal the identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Authors' contributions
SB conceived the idea; YNK collected and organized the literature and wrote the initial draft; SB wrote the final draft of the article. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
| 1. | Kumari O, Singh V. Prevalence and pattern of congenital musculoskeletal anomalies: A single centre study. J Clin Diag Res 2018;12:QC16-9. [Google Scholar] |
| 2. | Jacobsen ST, Crawford AH. Congenital vertical talus. J Pediatr Orthop 1983;3:306-10. [Google Scholar] |
| 3. | Seimon LP. Surgical correction of congenital vertical talus under the age of 2 years. J Pediatr Orthop 1987;7:405-11. [Google Scholar] |
| 4. | Coleman SS, Stelling FH 3rd, Jarrett J. Pathomechanics and treatment of congenital vertical talus. Clin Orthop Relat Res 1970;70:62-72. [Google Scholar] |
| 5. | McKie J, Radomisli T. Congenital vertical talus: A review. Clin Podiatr Med Surg 2010;27:145-56. [Google Scholar] |
| 6. | Hart ES, Grottkau BE, Rebello GN, Albright MB. The newborn foot: Diagnosis and management of common conditions. Orthop Nurs 2005;24:313-21. [Google Scholar] |
| 7. | Dobbs MB, Schoenecker PL, Gordon JE. Autosomal dominant transmission of isolated congenital vertical talus. Iowa Orthop J 2002;22:25-7. [Google Scholar] |
| 8. | Muhsin E. Common anomalies associated to congenital vertical talus: A single center experience. Int J Foot Ankle 2018;2:13. [Google Scholar] |
| 9. | Green NE, Lowery ER, Thomas R. Orthopaedic aspects of prune belly syndrome. J Pediatr Orthop 1993;13:496-501. [Google Scholar] |
| 10. | Krakowiak PA, Bohnsack JF, Carey JC, Bamshad M. Clinical analysis of a variant of Freeman-Sheldon syndrome (DA2B). Am J Med Genet 1998;76:93-8. [Google Scholar] |
| 11. | Haberlandt E, Löffler J, Hirst-Stadlmann A, Stöckl B, Judmaier W, Fischer H, et al. Split hand/split foot malformation associated with sensorineural deafness, inner and middle ear malformation, hypodontia, congenital vertical talus, and deletion of eight microsatellite markers in 7q21.1-q21.3. J Med Genet 2001;38:405-9. [Google Scholar] |
| 12. | Yalçin S, Kocaoglu B, Berker N, Erol B. Talectomy for the treatment of neglected pes equinovarus deformity in patients with neuromuscular involvement. Acta Orthop Traumatol Turc 2005;39:316-21. [Google Scholar] |
| 13. | Toydemir RM, Bamshad MJ. Sheldon-Hall syndrome. Orphanet J Rare Dis 2009;4:11. [Google Scholar] |
| 14. | Zhao N, Jiang M, Han W, Bian C, Li X, Huang F, et al. A novel mutation in TNNT3 associated with Sheldon-Hall syndrome in a Chinese family with vertical talus. Eur J Med Genet 2011;54:351-3. [Google Scholar] |
| 15. | Guo J, Wang L, Mo Z, Chen W, Fan Y. Biomechanical behavior of valgus foot in children with cerebral palsy: A comparative study. J Biomech 2015;48:3170-7. [Google Scholar] |
| 16. | Vorster AA, Beighton P, Ramesar RS. Digitotalar dysmorphism: Molecular elucidation. S Afr Med J 2016;106:253-5. [Google Scholar] |
| 17. | Rubio EI, Mehta N, Blask AR, Bulas DI. Prenatal congenital vertical talus (rocker bottom foot): A marker for multisystem anomalies. Pediatr Radiol 2017;47:1793-9. [Google Scholar] |
| 18. | Cho BC, Lee IH, Chung CY, Sung KH, Lee KM, Kwon SS, et al. Undercorrection of planovalgus deformity after calcaneal lengthening in patients with cerebral palsy. J Pediatr Orthop B 2018;27:206-13. [Google Scholar] |
| 19. | Drennan JC, Sharrard WJ. The pathological anatomy of convex pes valgus. J Bone Joint Surg Br 1971;53:455-61. [Google Scholar] |
| 20. | Ellis JN, Scheer GE. Congenital convex pes valgus. Clin Orthop Relat Res 1974;???:168-74. [Google Scholar] |
| 21. | Tachdjian MO. Congenital convex pes valgus. Orthop Clin North Am 1972;3:131-48. [Google Scholar] |
| 22. | Alaee F, Boehm S, Dobbs MB. A new approach to the treatment of congenital vertical talus. J Child Orthop 2007;1:165-74. [Google Scholar] |
| 23. | Patterson WR, Fitz DA, Smith WS. The pathologic anatomy of congenital convex pes valgus. Post mortem study of a newborn infant with bilateral involvement. J Bone Joint Surg Am 1968;50:458-66. [Google Scholar] |
| 24. | Specht EE. Congenital paralytic vertical talus. An anatomical study. J Bone Joint Surg Am 1975;57:842-7. [Google Scholar] |
| 25. | Drennan JC. Congenital vertical talus. Instr Course Lect 1996;45:315-22. [Google Scholar] |
| 26. | Dobbs MB, Purcell DB, Nunley R, Morcuende JA. Early results of a new method of treatment for idiopathic congenital vertical talus. J Bone Joint Surg Am 2006;88:1192-200. [Google Scholar] |
| 27. | Walker AP, Ghali NN, Silk FF. Congenital vertical talus. The results of staged operative reduction. J Bone Joint Surg Br 1985;67:117-21. [Google Scholar] |
| 28. | Stricker SJ, Rosen E. Early one-stage reconstruction of congenital vertical talus. Foot Ankle Int 1997;18:535-43. [Google Scholar] |
| 29. | Dobbs MB, Purcell DB, Nunley R, Morcuende JA. Early results of a new method of treatment for idiopathic congenital vertical talus. Surgical technique. J Bone Joint Surg Am 2007;89 Suppl 2 Pt. 1:111-21. [Google Scholar] |
| 30. | Ogata K, Schoenecker PL, Sheridan J. Congenital vertical talus and its familial occurrence: An analysis of 36 patients. Clin Orthop Relat Res 1979;139:128-32. [Google Scholar] |
| 31. | Hamanishi C. Congenital vertical talus: Classification with 69 cases and new measurement system. J Pediatr Orthop 1984;4:318-26. [Google Scholar] |
| 32. | Stern HJ, Clark RD, Stroberg AJ, Shohat M. Autosomal dominant transmission of isolated congenital vertical talus. Clin Genet 1989;36:427-30. [Google Scholar] |
| 33. | Levinsohn EM, Shrimpton AE, Cady RB, Packard DS, Hootnick DR. Congenital vertical talus in four generations of the same family. Skeletal Radiol 2004;33:649-54. [Google Scholar] |
| 34. | Dobbs MB, Gurnett CA, Pierce B, Exner GU, Robarge J, Morcuende JA, et al. HOXD10 M319K mutation in a family with isolated congenital vertical talus. J Orthop Res 2006;24:448-53. [Google Scholar] |
| 35. | Thomas JT, Kilpatrick MW, Lin K, Erlacher L, Lembessis P, Costa T, et al. Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet 1997;17:58-64. [Google Scholar] |
| 36. | Luyten FP. Cartilage-derived morphogenetic protein-1. Int J Biochem Cell Biol 1997;29:1241-4. [Google Scholar] |
| 37. | Faiyaz-Ul-Haque M, Ahmad W, Wahab A, Haque S, Azim AC, Zaidi SH, et al. Frameshift mutation in the cartilage-derived morphogenetic protein 1 (CDMP1) gene and severe acromesomelic chondrodysplasia resembling Grebe-type chondrodysplasia. Am J Med Genet 2002;111:31-7. [Google Scholar] |
| 38. | Shum L, Coleman CM, Hatakeyama Y, Tuan RS. Morphogenesis and dysmorphogenesis of the appendicular skeleton. Birth Defects Res C Embryo Today 2003;69:102-22. [Google Scholar] |
| 39. | Umair M, Rafique A, Ullah A, Ahmad F, Ali RH, Nasir A, et al. Novel homozygous sequence variants in the GDF5 gene underlie acromesomelic dysplasia type-grebe in consanguineous families. Congenit Anom (Kyoto) 2017;57:45-51. [Google Scholar] |
| 40. | Mumtaz S, Riaz HF, Touseef M, Basit S, Faiyaz Ul Haque M, Malik S. Recurrent mutation in CDMP1 in a family with Grebe chondrodysplasia: Broadening the phenotypic manifestation of syndrome in Pakistani population. Pak J Med Sci 2015;31:1542-4. [Google Scholar] |
| 41. | Al-Qattan MM, Al-Motairi MI, Al Balwi MA. Two novel homozygous missense mutations in the GDF5 gene cause brachydactyly type C. Am J Med Genet A 2015;167:1621-6. [Google Scholar] |
| 42. | Basit S, Naqvi SK, Wasif N, Ali G, Ansar M, Ahmad W. A novel insertion mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene underlies Grebe-type chondrodysplasia in a consanguineous Pakistani family. BMC Med Genet 2008;9:102. [Google Scholar] |
| 43. | Faiyaz-Ul-Haque M, Ahmad W, Zaidi SH, Haque S, Teebi AS, Ahmad M, et al. Mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene in a kindred affected with fibular hypoplasia and complex brachydactyly (DuPan syndrome). Clin Genet 2002;61:454-8. [Google Scholar] |
| 44. | Everman DB, Bartels CF, Yang Y, Yanamandra N, Goodman FR, Mendoza-Londono JR, et al. The mutational spectrum of brachydactyly type C. Am J Med Genet 2002;112:291-6. [Google Scholar] |
| 45. | Thomas JT, Lin K, Nandedkar M, Camargo M, Cervenka J, Luyten FP. A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat Genet 1996;12:315-7. [Google Scholar] |
| 46. | Polinkovsky A, Robin NH, Thomas JT, Irons M, Lynn A, Goodman FR, et al. Mutations in CDMP1 cause autosomal dominant brachydactyly type C. Nat Genet 1997;17:18-9. [Google Scholar] |
| 47. | Dobbs MB, Gurnett CA, Robarge J, Gordon JE, Morcuende JA, Bowcock AM. Variable hand and foot abnormalities in family with congenital vertical talus and CDMP-1 gene mutation. J Orthop Res 2005;23:1490-4. [Google Scholar] |
| 48. | Redline RW, Hudock P, MacFee M, Patterson P. Expression of AbdB-type homeobox genes in human tumors. Lab Invest 1994;71:663-70. [Google Scholar] |
| 49. | Zhang J, Liu S, Zhang D, Ma Z, Sun L. Homeobox D10, a tumor suppressor, inhibits the proliferation and migration of esophageal squamous cell carcinoma. J Cell Biochem 2019;120:13717-25. [Google Scholar] |
| 50. | Shrimpton AE, Levinsohn EM, Yozawitz JM, Packard DS Jr., Cady RB, Middleton FA, et al. A HOX gene mutation in a family with isolated congenital vertical talus and Charcot-Marie-Tooth disease. Am J Hum Genet 2004;75:92-6. [Google Scholar] |
| 51. | Alvarado DM, McCall K, Hecht JT, Dobbs MB, Gurnett CA. Deletions of 5' HOXC genes are associated with lower extremity malformations, including clubfoot and vertical talus. J Med Genet 2016;53:250-5. [Google Scholar] |
| 52. | Gurnett CA, Keppel C, Bick J, Bowcock AM, Dobbs MB. Absence of HOXD10 mutations in idiopathic clubfoot and sporadic vertical talus. Clin Orthop Relat Res 2007;462:27-31. [Google Scholar] |
| 53. | Wilson MG, Towner JW, Forsman I, Siris E. Syndromes associated with deletion of the long arm of chromosome 18[del (18q)]. Am J Med Genet 1979;3:155-74. [Google Scholar] |
| 54. | Gay CT, Hardies LJ, Rauch RA, Lancaster JL, Plaetke R, DuPont BR, et al. Magnetic resonance imaging demonstrates incomplete myelination in 18q- syndrome: Evidence for myelin basic protein haploinsufficiency. Am J Med Genet 1997;74:422-31. [Google Scholar] |
| 55. | Kline AD, White ME, Wapner R, Rojas K, Biesecker LG, Kamholz J, et al. Molecular analysis of the 18q- syndrome—and correlation with phenotype. Am J Hum Genet 1993;52:895-906. [Google Scholar] |
| 56. | Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, et al. Congenital anomalies and anthropometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet 1999;85:455-62. [Google Scholar] |
| 57. | Veltman JA, Jonkers Y, Nuijten I, Janssen I, van der Vliet W, Huys E, et al. Definition of a critical region on chromosome 18 for congenital aural atresia by arrayCGH. Am J Hum Genet 2003;72:1578-84. [Google Scholar] |
| 58. | Dostal A, Nemeckova J, Gaillyova R, Vranova V, Zezulkova D, Lejska M, et al. Identification of 2.3-Mb gene locus for congenital aural atresia in 18q22.3 deletion: A case report analyzed by comparative genomic hybridization. Otol Neurotol 2006;27:427-32. [Google Scholar] |
| 59. | Dostal A, Nemeckova J, Gaillyova R. The 18q deletion syndrome and analysis of the critical region for orofacial cleft at 18q22.3. J Craniomaxillofac Surg 2009;37:272-5. [Google Scholar] |
| 60. | Feenstra I, Vissers LE, Orsel M, van Kessel AG, Brunner HG, Veltman JA, et al. Genotype-phenotype mapping of chromosome 18q deletions by high-resolution array CGH: An update of the phenotypic map. Am J Med Genet A 2007;143A: 1858-67. [Google Scholar] |
| 61. | Cody JD, Heard PL, Crandall AC, Carter EM, Li J, Hardies LJ, et al. Narrowing critical regions and determining penetrance for selected 18q- phenotypes. Am J Med Genet A 2009;149A: 1421-30. [Google Scholar] |
| 62. | Mark PR, Radlinski BC, Core N, Fryer A, Kirk EP, Haldeman-Englert CR. Narrowing the critical region for congenital vertical talus in patients with interstitial 18q deletions. Am J Med Genet A 2013;161A: 1117-21. [Google Scholar] |
| 63. | Alexandre E, Graba Y, Fasano L, Gallet A, Perrin L, De Zulueta P, et al. The drosophila teashirt homeotic protein is a DNA-binding protein and modulo, a HOM-C regulated modifier of variegation, is a likely candidate for being a direct target gene. Mech Dev 1996;59:191-204. [Google Scholar] |
| 64. | Coré N, Caubit X, Metchat A, Boned A, Djabali M, Fasano L. Tshz1 is required for axial skeleton, soft palate and middle ear development in mice. Dev Biol 2007;308:407-20. [Google Scholar] |
| 65. | Tassano E, Severino M, Rosina S, Papa R, Tortora D, Gimelli G, et al. Interstitial de novo 18q22.3q23 deletion: Clinical, neuroradiological and molecular characterization of a new case and review of the literature. Mol Cytogenet 2016;9:78. [Google Scholar] |
| 66. | Merrill LJ, Gurnett CA, Connolly AM, Pestronk A, Dobbs MB. Skeletal muscle abnormalities and genetic factors related to vertical talus. Clin Orthop Relat Res 2011;469:1167-74. [Google Scholar] |
| 67. | Wang WB, Kong LC, Zuo RT, Kang QL. Identification of a novel pathogenic mutation of the MYH3 gene in a family with distal arthrogryposis type 2B. Mol Med Rep 2020;21:438-44. [Google Scholar] |
| 68. | Li S, You Y, Gao J, Mao B, Cao Y, Zhao X, et al. Novel mutations in TPM2 and PIEZO2 are responsible for distal arthrogryposis (DA) 2B and mild DA in two Chinese families. BMC Med Genet 2018;19:179. [Google Scholar] |
| 69. | Xu Y, Kang QL, Zhang ZL. A MYH3 mutation identified for the first time in a Chinese family with Sheldon-Hall syndrome (DA2B). Neuromuscul Disord 2018;28:456-62. [Google Scholar] |
| 70. | Čulić V, Miyake N, Janković S, Petrović D, Šimunović M, Đapić T, et al. Distal arthrogryposis with variable clinical expression caused by TNNI2 mutation. Hum Genome Var 2016;3:16035. [Google Scholar] |
| 71. | Beck AE, McMillin MJ, Gildersleeve HI, Kezele PR, Shively KM, Carey JC, et al. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am J Med Genet A 2013;161A:550-5. [Google Scholar] |
| 72. | Weymouth KS, Blanton SH, Bamshad MJ, Beck AE, Alvarez C, Richards S, et al. Variants in genes that encode muscle contractile proteins influence risk for isolated clubfoot. Am J Med Genet A 2011;155A:2170-9. [Google Scholar] |
| 73. | Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: A review and update. J Bone Joint Surg Am 2009;91 Suppl 4:40-6. [Google Scholar] |
| 74. | Davidson AE, Siddiqui FM, Lopez MA, Lunt P, Carlson HA, Moore BE, et al. Novel deletion of lysine 7 expands the clinical, histopathological and genetic spectrum of TPM2-related myopathies. Brain 2013;136:508-21. [Google Scholar] |
| 75. | Gurnett CA, Alaee F, Desruisseau D, Boehm S, Dobbs MB. Skeletal muscle contractile gene (TNNT3, MYH3, TPM2) mutations not found in vertical talus or clubfoot. Clin Orthop Relat Res 2009;467:1195-200. [Google Scholar] |
| 76. | Aroojis AJ, King MM, Donohoe M, Riddle EC, Kumar SJ. Congenital vertical talus in arthrogryposis and other contractural syndromes. Clin Orthop Relat Res 2005;434:26-32. [Google Scholar] |
| 77. | Angsanuntsukh C, Oto M, Holmes L, Rogers KJ, King MM, Donohoe M, et al. Congenital vertical talus in multiple pterygium syndrome. J Pediatr Orthop 2011;31:564-9. [Google Scholar] |
| 78. | Fornaciari P, Gilgen A, Zwicky L, Horn Lang T, Hintermann B. Isolated talonavicular fusion with tension band for Müller-Weiss syndrome. Foot Ankle Int 2014;35:1316-22. [Google Scholar] |
Fulltext Views
3,343
PDF downloads
349




