Translate this page into:
Rare painful pediatric deformity; when surgery does harm: Case report and review of the literature
2 Alameerat Private Hospital Kindi Street, Harthiya, Baghdad, Iraq
3 Department of Orthopedics, Ibri Regional Hospital, Al Dharia, Sultanate of Oman
4 Al-Mustansiriya University/College of Medicine, Neurosurgery Surgry, Baghdad, Iraq
Corresponding Author:
Luay M Al-Naser
Department of Orthopedics, Baquba Teaching Hospital, Diyala
Iraq
luay.muayad.lm@gmail.com
| How to cite this article: Al-Naser LM, Hasan GA, Sheta RA, Mazin YA, Raheem HQ. Rare painful pediatric deformity; when surgery does harm: Case report and review of the literature. J Musculoskelet Surg Res 2019;3:307-310 |
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a connective tissue disease that is extremely rare. Fibrous tissue in muscles, tendons, and ligaments get ossified spontaneously or damaged by a mutation of the body's repair mechanism. In this report, we present a case of FOP, which is considered one of the rarest diseases that can cause scoliosis and new bone formation around the spine and almost everywhere in the body. A 14-year-old female presented with scoliosis and limitation in spine and upper limb movements. The condition started at the age of 9 years with a painful mass in the right side of the upper back. At that time, the parents asked for medical help; a general surgeon advised and performed an excisional biopsy of the mass. Subsequently, the mass reappeared and continued to grow, covering nearly the entire back. Radiological investigations revealed a well-formed bony band extending from the right shoulder to the iliac bone posteriorly with bony formations in the left shoulder, back, medial aspects of the arms, axilla, and buttocks. FOP is an extremely challenging disorder with difficulties in both diagnosis and management, and large malformed toes with well-formed ossification anywhere in the body should raise the suspicion of the diagnosis. Any surgical intervention, including biopsy, will harm the patient and should be avoided.Introduction
Fibrodysplasia ossificans progressiva (FOP) begins in childhood as painful, erythematous subfascial nodules, most commonly located on the posterior neck and back. It is a rare, autosomal-dominant, progressive, disabling disorder affecting the musculoskeletal system.[1] The exact pathophysiology of FOP is still unknown.[2] The condition is multifocal, induced usually after trauma. The prevalence of FOP is about 1 case per 1.64 million in the United Kingdom. While in France, it has been calculated as 1.36 per million inhabitants.[3] Fewer than 200 cases have been described worldwide, and it is more common in females than in males. FOP usually starts in early infancy. The prognosis is poor because of the involvement of the thoracic muscles and leading to restrictive lung disease.[4]
The main target of ectopic bone formation is usually involving the axial muscles, but it could occur in the ligaments, fascia, aponeuroses, tendons, and joint capsules.[5] Due to ankylosis of the spine and ribs, the movement is restricted in all directions. Maxillofacial region involvement also has been described.[6] It may produce a fusion between the mandibular ramus and the zygomatic complex and trismus. Other characteristic features seen in some cases are alopecia and deafness.[7] Surgical trauma can lead to further aggravation of the condition, even in diagnostic biopsies, that is why inexperienced surgeon might face problems in the management of such cases, and try to avoid any surgical interventions whenever is possible even biopsies FOP.[8] While the characteristic features are bony masses within muscles, which are best diagnosed by computed tomography (CT) scan, even in the early stages.[9] There is no definitive treatment; however, steroid and bisphosphonate might be used during flare-ups, but still no evidence of its positive effect. Gene therapy may hold promise in FOP treatment.
The aim of our report is to present a rare cause of painful pediatric spine deformity with a previous surgical intervention that caused progression of the disease and to deliver a message that any surgical intervention in such a case will do harm rather than help.
Case Report
A 14-year-old female presented with scoliosis, and limitation in bending and movements, especially the upper limb movements. The condition started when she was 9 years old with a painful mass in the upper right back. After medical consultation, a general surgeon advised and performed an excisional biopsy of the mass, which appeared benign [Figure - 1]. There was a recurrent, progressive mass that involved the entire back. There was progressive deterioration in forward and side bending and inability to abduct the right shoulder, which started 1 year after the biopsy and progressed until the time of presentation.
![[Figure - 1]](#fig_SaudiOrthopJ_2019_3_3_307_263654_f1.jpg){kind=link}
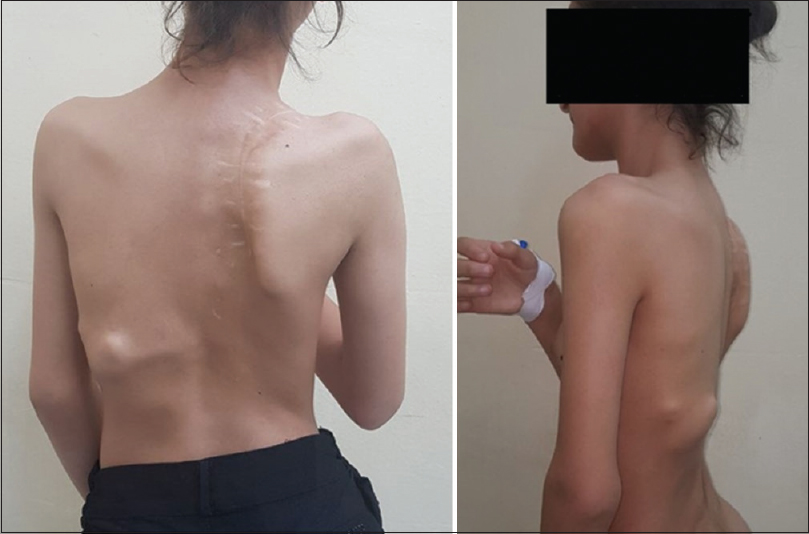 |
| Figure 1: Multiple hard irregular masses in the deformed back |
She has continued her schooling and has done well, leading a relatively normal life with deformity and occasional pain. She did not complain of respiratory problems, has no conductive hearing defects, no retinopathy, and no childhood glaucoma. She had normal teeth development and did not start menstruating. She has three normal younger brothers, and there was no family history for the same condition. On examination, we noticed malformed hypoplastic bilateral big toes [Figure - 2], which was present since birth. Furthermore, a grossly deformed back with a hard, irregular, well-formed, painless mass extending from the right shoulder posteriorly down to the right pelvic area, along with other smaller hard masses on the left side [Figure - 1]. There was a severe limitation in all movements of the right upper limb and to a lesser degree in the left upper limb. She was not anemic, and the alkaline phosphatase was elevated (204.49 U/L, the normal reference for females is 55–104 U/L). Chromosomal microarray did not identify any abnormality, and no further genetic study was performed on either the patient or her family, as this option was not available in our country at the time. Radiological investigations (radiographs) revealed soft tissue ossification and calcification involving the back and right side of the neck, forming a band-like structure from the C3 down to L5, with fusion of the right clavicle and right scapula and it also showed dextrocardia [Figure - 3]. The pelvis radiograph showed bilateral coxa valga [Figure - 4]. Knee radiographs revealed distal femur osteochondromas [Figure - 4]. The 3D CT scan of the entire spine showed in detail the ossified band that extended from C3 to L5 with fusion of the clavicle in the right side, and another ossified band extending from D12 to the sacrum and right-sided dorsal scoliosis [Figure - 5]. No specific treatment was given to the patient (e.g., bisphosphonate) as there was no clear evidence that such a treatment would help. At the final follow-up, approximately 8-year postdiagnosis when the biopsy was performed, the patient showed mild progression with no further surgical intervention.
![[Figure - 2]](#fig_SaudiOrthopJ_2019_3_3_307_263654_f2.jpg){kind=link}
![[Figure - 3]](#fig_SaudiOrthopJ_2019_3_3_307_263654_f3.jpg){kind=link}
![[Figure - 4]](#fig_SaudiOrthopJ_2019_3_3_307_263654_f4.jpg){kind=link}
![[Figure - 5]](#fig_SaudiOrthopJ_2019_3_3_307_263654_f5.jpg){kind=link}
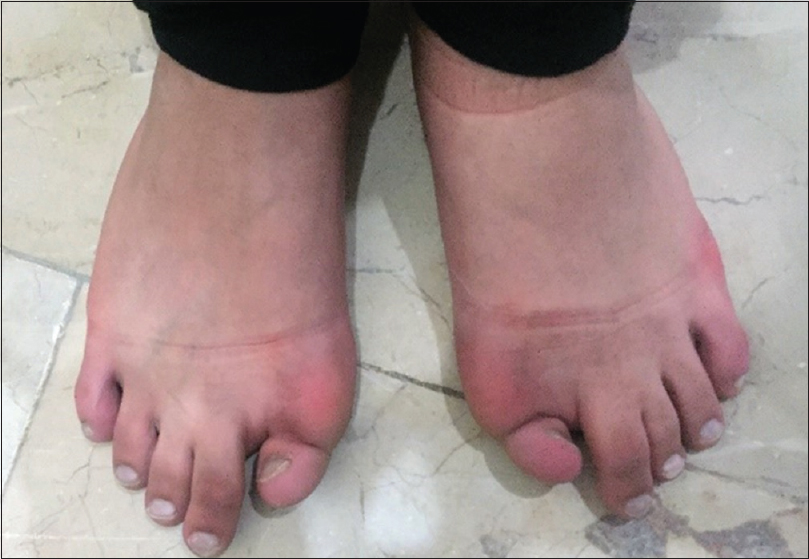 |
| Figure 2: Malformed hypoplastic bilateral big toes |
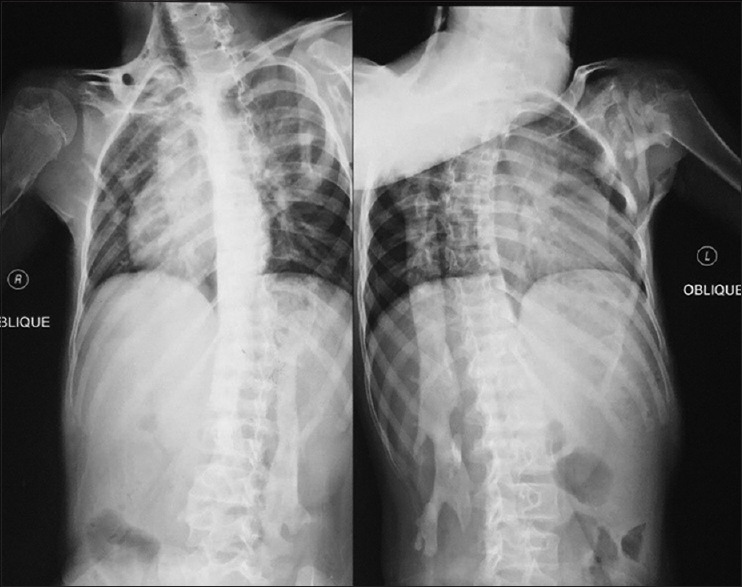 |
| Figure 3: Anteroposterior radiograph of the spine showing the right-sided radiopaque band extending from the cervical to the lumbar spine, and also showing dextrocardia |
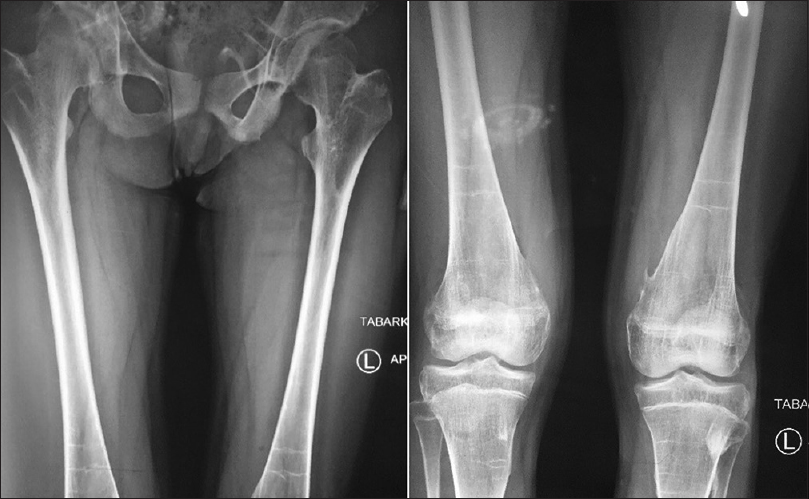 |
| Figure 4: Radiograph of the pelvis and both femurs, showing bilateral coxa valga- Anteroposterior radiograph of both knees, which is showing multiple distal femur osteochondromas |
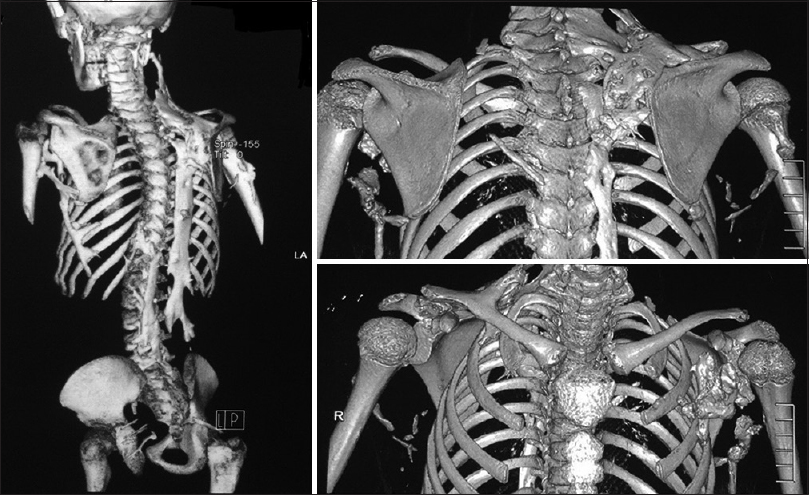 |
| Figure 5: Computed tomography scan of the spine, with three-dimensional reconstruction showing the ossified band that extends from the cervical to the lumbar spine |
Discussion
FOP is a rare autosomal dominant inherited disorder of connective tissue without apparent racial, ethnic, or geographical variation.[10] FOP is a disabling disease characterized by an excessive formation of endochondral bone within connective tissues.[11] Our case is more likely due to a sporadic spontaneous mutation, as there is no positive family history for the same condition. Our patient is 14 years old, and the disease first appeared when she was 9 years old as a painless, progressive mass on the right side of the upper back. As the condition is rare, and the family consulted a general surgeon at that time, the surgeon performed an excisional biopsy and the result appeared as a benign lesion. Subsequently, the lesion recurred and multiplied with progressive associated back deformities, which were palpable on examination.
In a review of the literature, no surgical intervention is recommended, including biopsy, as it will cause a flare-up of the disease and will lead to disease progression.[12] Hence, the cause of the flare-up and progression in our case is most likely due to the previous excisional biopsy that the patient underwent 5 years before presentation, as the biopsy actually harmed the patient.
Other characteristic features of the disease are present in our case, including malformed hypoplastic bilateral big toes, which is regarded as one of the characteristics and diagnostic features of FOP,[13] the distal femur osteochondroma, that was present in the lateral aspect of distal femur, as shown in [Figure - 3]. In the review of the literature, Deirmengian et al. found that osteochondromas were located in proximal tibia.[5] Painful back deformities are due to ossification of the back muscles and the fusion band. This is another characteristic feature of FOP, which appears as multiple soft, rubbery, or hard masses, depending on whether there is ossification within the mass or not. The ossified band and masses will cause spine deformities, which was evident in our case, as painful scoliosis is due to the ossified band that extended from the cervical spine to the sacrum.[13]
The laboratory studies (liver and renal function, coagulation profile, and complete blood picture) in our case were normal, except for elevated alkaline phosphatase (204.49 U/L), which is double the normal range for females. This increase in the level is characteristic for children and is a natural effect of the physiologic growth of bones.[13] The chromosomal study of the case was negative. While the specific mutations in the gene encoding ACVR1, a BMP type I receptor, were unavailable, and we did not perform them in our case, as the clinical and radiological features were characteristic and gene-specific studies are not available in our hospital. Even without a genetic study, in our case, we could reach the diagnosis based on the clinical presentation and progression of symptoms. In the review of the literature, there is no specific recommendation for a specific treatment, and follow-up is recommended in cases such as ours. Patients should be observed, and the cardiorespiratory system should be monitored.[4] According to the natural history of the case presentation with the presence of other characteristic features including the osteochondroma around the knee and hallux deformities and the laboratory finding with radiological confirmation including radiographs, CT scan, and magnetic resonance imaging, the differential diagnosis of osteosarcoma was excluded.
Conclusion
FOP is an extremely challenging disorder with difficulties in both diagnosis and management. It presents with malformed big toes and well-formed ossification presenting anywhere in the body should raise the suspicion of it. Surgical intervention, even a biopsy, will harm the patient and should be avoided.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Authors' contribution
LMN, GAH, RAS, YAM, HQR are equally conceived and designed the study, conducted the research, provided the research materials, and collected and organized, analyzed and interpreted data. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
| 1. | Feldman G, Li M, Martin S, Urbanek M, Urtizberea JA, Fardeau M, et al. Fibrodysplasia ossificans progressiva, a heritable disorder of severe heterotopic ossification, maps to human chromosome 4q27-31. Am J Hum Genet 2000;66:128-35. [Google Scholar] |
| 2. | de la Peña LS, Billings PC, Fiori JL, Ahn J, Kaplan FS, Shore EM. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res 2005;20:1168-76. [Google Scholar] |
| 3. | Baujat G, Choquet R, Bouée S, Jeanbat V, Courouve L, Ruel A, et al. Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: An estimate based on a record linkage of two national databases. Orphanet J Rare Dis 2017;12:123. [Google Scholar] |
| 4. | Kaplan FS, Zasloff MA, Kitterman JA, Shore EM, Hong CC, Rocke DM. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am 2010;92:686-91. [Google Scholar] |
| 5. | Deirmengian GK, Hebela NM, O'Connell M, Glaser DL, Shore EM, Kaplan FS. Proximal tibial osteochondromas in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am 2008;90:366-74. [Google Scholar] |
| 6. | Karakasis D, Triantafyllidou E, Kavadia S. Extra-articular ankylosis of the coronoid processes to the base of the skull. A case report. J Craniomaxillofac Surg 1989;17:46-9. [Google Scholar] |
| 7. | Levy CE, Lash AT, Janoff HB, Kaplan FS. Conductive hearing loss in individuals with fibrodysplasia ossificans progressiva. Am J Audiol 1999;8:29-33. [Google Scholar] |
| 8. | Muthu V, Khaire NS, Varma S. Fibrodysplasia ossificans progressiva – Primum non nocere. Clin Rheumatol 2014;33:591-2. [Google Scholar] |
| 9. | Chuang TL, Ho KW, Wang YF. A bizarre bone scan of fibrodysplasia ossificans progressiva. Clin Nucl Med 2018;43:433-5. [Google Scholar] |
| 10. | Hüning I, Gillessen-Kaesbach G. Fibrodysplasia ossificans progressiva: Clinical course, genetic mutations and genotype-phenotype correlation. Mol Syndromol 2014;5:201-11. [Google Scholar] |
| 11. | Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006;38:525-7. [Google Scholar] |
| 12. | Kaplan FS, Glaser DL, Pignolo RJ, Shore EM. A new era for fibrodysplasia ossificans progressiva: A druggable target for the second skeleton. Expert Opin Biol Ther 2007;7:705-12. [Google Scholar] |
| 13. | Pignolo RJ, Baujat G, Brown MA, De Cunto C, DiRocco M, Hsiao EC, et al. Natural history of fibrodysplasia ossificans progressiva: cross-sectional analysis of annotated baseline phenotypes. Orphanet J Rare Dis. 2019;14:98. [Google Scholar] |
Fulltext Views
1,532
PDF downloads
511




