Translate this page into:
A systematized approach to radiographic assessment of commonly seen genetic bone diseases in children: A pictorial review
2 Department of Paediatrics, Genetics Unit, Faculty of Medicine, Ain Shams University, Abbassia, Cairo, Egypt
3 Department of Radiology, Faculty of Medicine, Ain Shams University, Abbassia, Cairo, Egypt
4 Center of Medical Genetics, Faculty of Medicine, Ain Shams University, Abbassia, Cairo, Egypt
Corresponding Author:
Tamer A El-Sobky
Department of Orthopaedic Surgery, Division of Paediatric Orthopaedics, Faculty of Medicine, Ain-Shams University, Abbassia, Cairo
Egypt
tamersh@hotmail.comHossam M Sakr
Department of Radiology, Faculty of Medicine, Ain Shams University, Abbassia, Cairo
Egypt
drhossamsakr@yahoo.com
| How to cite this article: El-Sobky TA, Shawky RM, Sakr HM, Elsayed SM, Elsayed NS, Ragheb SG, Gamal R. A systematized approach to radiographic assessment of commonly seen genetic bone diseases in children: A pictorial review. J Musculoskelet Surg Res 2017;1:25-32 |
Abstract
Genetic skeletal dysplasias or osteochondrodysplasias constitute a large group of hereditable disorders with generalized skeletal involvement that can cause significant morbidity and mortality. Although most genetic skeletal dysplasias have been well identified clinically, the overlap between dysplasia subtypes may create diagnostic challenges. The plain radiographic presentation of genetic skeletal dysplasias may closely mimic rheumatologic, hematologic, and Perthes disease. Accurate review of the plain radiographic and clinical data may allow to prioritize and conduct gene sequencing tests efficiently. Bone imaging plays a distinctive role in diagnosis. We reviewed the radiologic profile of patients with some commonly seen skeletal dysplasias; achondroplasia, multiple epiphyseal dysplasia, pseudoachondroplasia, spondyloepiphyseal dysplasia tarda, osteogenesis imperfecta, mucopolysaccharidosis, and spondylometaphyseal and spondyloepimetaphyseal dysplasia. We proposed a specific radiographic approach to the differential diagnosis of commonly seen osteochondrodysplasias. The pictorial essay demonstrates that careful plain radiographic evaluation can be a very beneficial tool to the diagnostic process of osteochondrodysplasias.
Study Highlights
- Skeletal radiography can deliver significant clues to the diagnosis of commonly seen osteochondrodysplasias
- Adopting a systematized approach to the radiographic assessment of osteochondrodysplasias can optimize the role of diagnostic imaging
- Meticulous reporting of the pattern, extent, and severity of radiographic skeletal pathology is an essential part of the diagnostic process
- Radioclinical correlations are indispensable to accurate diagnosis of commonly seen osteochondrodysplasias.
Introduction
Genetic skeletal dysplasias or osteochondrodysplasias comprise a large group of clinically and radiologically diverse disorders that stem from disordered growth and development of bone and cartilage. More than 450 well-characterized osteochondrodysplasias have been recognized. Despite the relative rarity of each type, the summed incidence of these osteochondrodysplasias is not trivial. This imperfect osseocartilaginous biology is usually attributed to defects in structural proteins, metabolic processes, or growth plate regulation.[1],[2],[3],[4] Arriving at a definitive diagnosis is critical in understanding prognosis, genetic counseling, preventing complications, and timely institution of available medical and surgical management.[5],[6],[7],[8],[9],[10],[11] Skeletal dysplasias can result in functional disability secondary to joint destruction, limb deformities, neurologic involvement, and reduced health-related quality of life. Extraskeletal complications can occur in association with osteochondrodysplasia.[12],[13],[14],[15],[16],[17],[18] Although most osteochondrodysplasias have been well identified clinically, the overlap between dysplasia subtypes may create diagnostic challenges.[19],[20],[21],[22] Clinical overlaps between osteochondrodysplasias and rheumatologic disorders have been reported.[23] Plain radiographic imaging remains a substantial contributor to accurate diagnosis of skeletal dysplasias in childhood.[24],[25],[26],[27],[28] Accurate review of the plain radiographic and clinical data may allow to prioritize and conduct gene sequencing tests efficiently.[26],[27] The goal of this study is to familiarize radiologists, pediatricians, and orthopedists with the typical imaging features of some of the commonly encountered osteochondrodysplasias and their differential diagnoses. The radiologic characteristics of osteochondrodysplasias were reviewed through adopting a specific systematized approach. The diagnosis was based on the coexistence of classic radioclinical features, biochemical testing, and available molecular analysis.
Guidelines for Radiologic Evaluation
Clinical assessment is an integral part of the diagnostic process. Anthropometric measurements are of particular significance for determining the type of disproportionate short-stature, short-limb versus short-trunk. The morphologic examination can provide valuable clues to the differential diagnosis. Detailed neurological and thoracoabdominal examination may be dictated by the clinical scenario.[19],[29],[30],[31] Mucopolysaccharidosis among others carries a significant potential for cranial and spinal complications.[14],[15] Osteochondrodysplasias may exhibit a deceptive radiographic resemblance to rheumatologic,[32] hematologic,[33],[34] and Perthes disease.[35] Adopting a systematized approach to the radiographic assessment of osteochondrodysplasias is pivotal to an accurate diagnosis. We propose adherence to the following guidelines: (a) a complete whole-body skeletal survey including orthogonal views of the skull, spine, pelvis, and all extremities with separate views for the hands, (b) technically sound and optimally positioned radiographs, (c) a general overview of bone density, architecture, alignment, pathologic fractures, and eruption patterns of secondary ossification nuclei, (d) determination of the predominant skeletal involvement, spine/axial versus limb/appendicular affection, (e) determination of most affected segment by shortening in the limbs, rhizomelic (proximal segments), mesomelic (middle segments), acromelic (distal segments), micromelic (universal limb involvement), versus the most affected spine segment, (f) identification of pattern of affection of different components of bone geography, joint, epiphysis, physis, metaphysis, or diaphysis, and (g) determination of bilateralism and symmetry of lesions [Figure - 1].
![[Figure - 1]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f1.jpg){kind=link}
 |
| Figure 1: A simplified scheme for radiographic differential diagnosis of commonly seen osteochondrodysplasias in children |
Achondroplasia
Achondroplasia is the most common nonlethal osteochondrodysplasias. It is caused by heterozygous mutation in the FGFR3 gene on chromosome 4p16.3. Achondroplasia is inherited as an autosomal dominant with essentially complete penetrance.[2] Patients demonstrate unique radioclinical features that are readily detectable at birth. Clinically, patients present with rhizomelic short-limb short stature particularly affecting the humeri and femora, macrocephaly, frontal bossing, midface hypoplasia, exaggerated lumbar lordosis, genu varum, and trident hand. There is a tendency to generalized joint laxity except for limitation of elbow extension and forearm rotation. In addition, patients exhibit predominant metaphyseal changes on radiographs [Figure - 2].[2],[3],[19]
![[Figure - 2]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f2.jpg){kind=link}
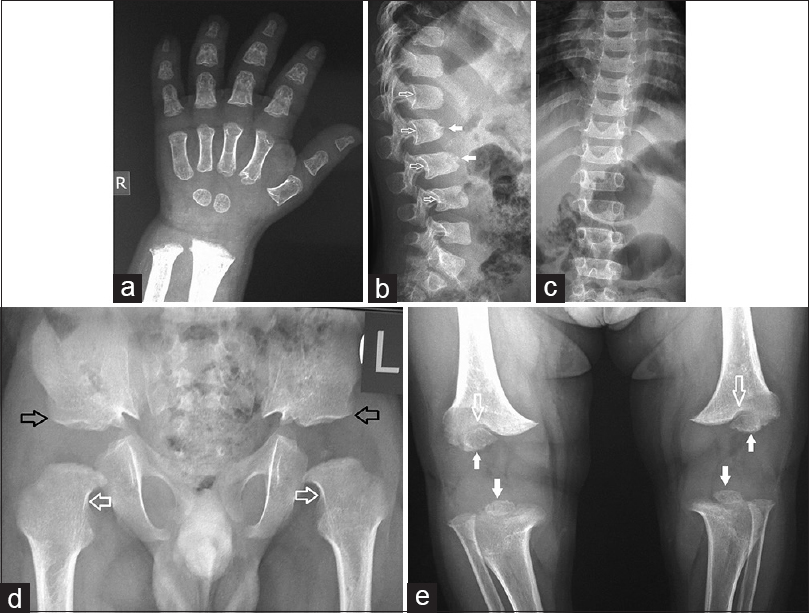 |
| Figure 2: (a-e) Achondroplasia, 2-year-old boy. Hand radiograph anteroposterior view (a) showing a spade-like hand with clear separation of fingers and uniform length of the 2nd, 3rd, 4th fingers and short phalanges with metaphyseal widening. Dorsolumbar spine radiograph lateral view (b) shows dysplastic bullet-shaped vertebrae with anterior beaking (solid arrows), posterior scalloping of vertebral bodies (hollow arrows), and mild dorsal kyphosis. Note the shortened ribs. Dorsolumbar spine radiograph anteroposterior view (c) shows an unchanged interpedicular distance caudally, especially in the lumbar segments. In contrast, pseudoachondroplasia patients exhibit normal widening of the interpedicular distances caudally. This is an important distinguishing radiographic feature. Pelvic radiograph anteroposterior view (d) demonstrates short broad femoral necks (white arrows), flat horizontal acetabuli (black arrows), and dysplastic iliac bones with squaring and rounding of corners. The proximal femoral epiphyses are totally and partially unossified in the right and left hips, respectively. Note that abnormalities are bilateral and symmetric. Knee radiograph anteroposterior view (e) demonstrates metaphyseal flaring and cupping of the distal femora and proximal tibias with relatively unaffected epiphyseal size (solid arrows). Note the characteristic V-shaped configuration of the distal femoral metaphyses (hollow arrows) producing a ball-in-socket epiphysis. Note the bilateral and symmetric lesions. Mark the generally intact morphology and texture of epiphyses (d and e) |
Multiple epiphyseal dysplasia and pseudoachondroplasia
Multiple epiphyseal dysplasia (MED) and pseudoachondroplasia are commonly encountered osteochondrodysplasias. MED is a heterogeneous disorder caused by mutations in several genes that have originated in various genotypic-phenotypic correlations. MED1 is caused by a heterozygous mutation in the gene encoding cartilage oligomeric matrix protein (COMP). Mutation in the COMP gene can also cause a clinically and radiologically similar but more extreme disorder pseudoachondroplasia. MED1 and pseudoachondroplasia are inherited in an autosomal dominant pattern and are collectively referred to as COMP-related osteochondrodysplasias.[2],[26],[27] Patients with MED usually manifest in early-to-mid childhood with deteriorating joint pain and stiffness and tend to develop a milder form of short stature. Pseudoachondroplasia usually manifests in the 2nd year of life with ligamentous laxity and tends to develop severe disproportionate short stature. Patients with MED and pseudoachondroplasia usually develop progressive degenerative joint disease often necessitating a hip joint replacement in early adulthood. In contrast to pseudoachondroplasia, spinal abnormalities in MED are usually less pronounced or absent.[21],[26],[27],[31],[36] The imaging features of MED and pseudoachondroplasia are depicted in [Figure - 3] and [Figure - 4].
![[Figure - 3]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f3.jpg){kind=link}
![[Figure - 4]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f4.jpg){kind=link}
 |
| Figure 3: (a and b) Multiple epiphyseal dysplasia. Pelvis and hip radiograph anteroposterior view (a). Note the broad short necks and irregular eruption of proximal femoral epiphyses with considerable symmetry of lesions. The greater trochanteric apophyses are overriding with an irregular ossification pattern. Notice the undersized ossification nucleus of the proximal femora in contrast to the fully erupted ossification nucleus of the greater trochanter. Normally, the ossification nucleus of the proximal femur appears before that of the greater trochanter. Observe the acetabular widening with subchondral sclerosis and periarticular irregularities. Knee radiograph anteroposterior view (b). Observe the epiphyseal irregularities and mottling (coexistence of lytic and sclerotic lesions) around the knee, epiphyseal vertical defects (hollow arrows), rarefaction, and depression of the medial tibial metaphyses (solid arrows). Mark the characteristic bilateralism and symmetry of described lesions |
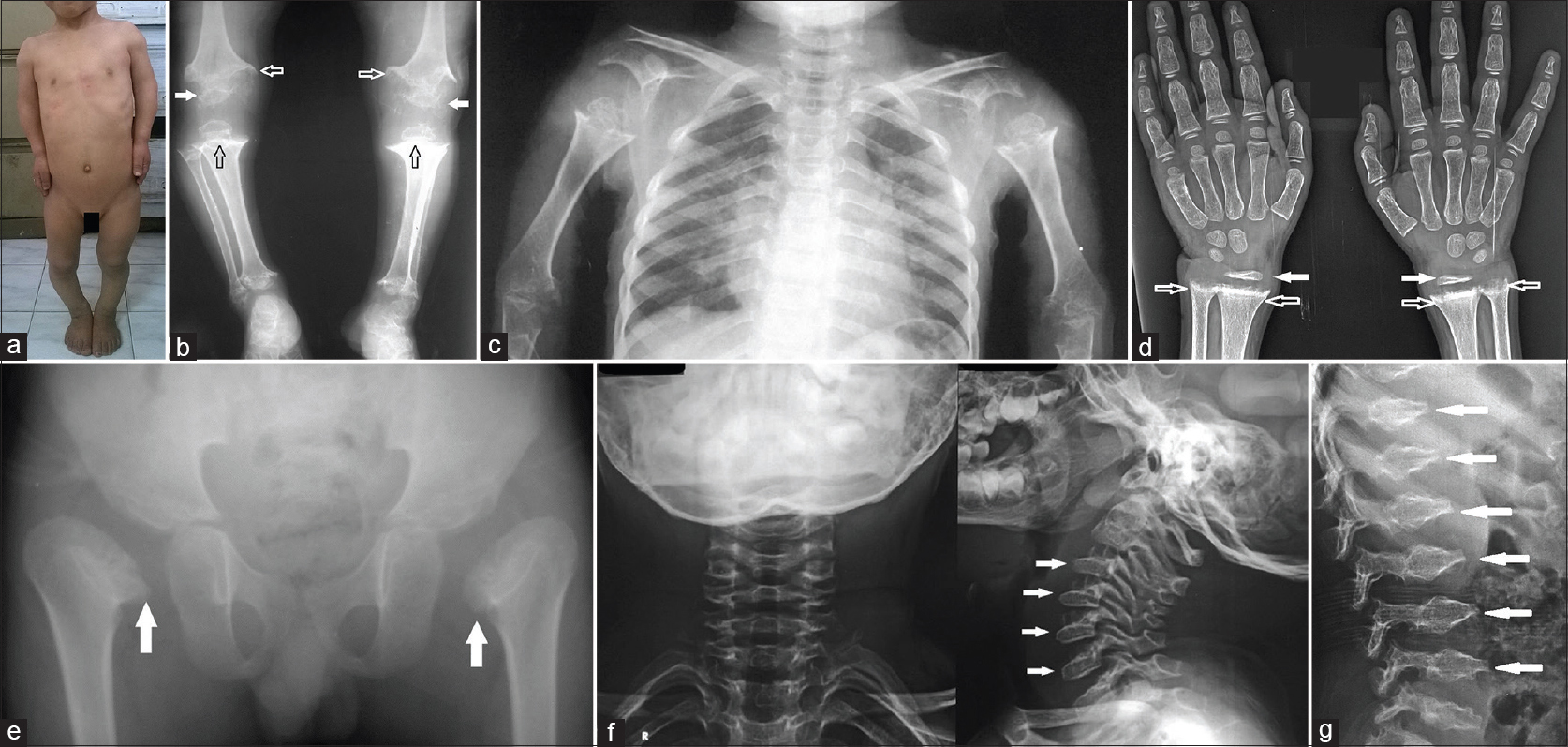 |
| Figure 4: (a-g) Pseudoachondroplasia. A 9-year-old female patient 1. Notice the disproportionate short-limb short stature. Mark the arm and thigh shortening (rhizomelic shortening), bow legs, and broadening of distal ends of forearm and tibias (a). Anteroposterior radiograph of both legs (b) demonstrating metaphyseal flaring and cupping (hollow white arrows), epiphyseal irregularities (solid white arrows), and dense sclerotic lines of the distal metaphyses (black arrows). All these changes are known as “rachitic-like changes”. Similar changes can be appreciated in the anteroposterior radiograph of both arms (c). A 9-year-old male with “hypophosphatemic rickets” patient 2. Hand and wrist radiographs (d) show typical rachitic changes in metaphyses of the distal radiuses and ulnae (hollow arrows). Note the small for age distal radial epiphyses (solid arrows). Notice the deceptive resemblance between these rachitic changes and the “rachitic-like changes” observed in the pseudoachondroplasia radiographs. Note the symmetry of epimetaphyseal lesions (b-d). Anteroposterior radiograph of the hips (e) of an 8-year-old male patient 3 demonstrates the absence of ossification in the proximal femoral epiphyses and greater trochanteric apophyses, the characteristic medial beaking of the neck (arrows) and coxa vara. A 4-year-old male patient 4. Cervical spine radiographs (f) depict the characteristic platyspondyly (uniformly reduced vertebral height) in anteroposterior view and atypical bullet-shaped/oval vertebrae (convexity of superior border with mild anterior and central beaking) in lateral view. Lumbar spine radiograph lateral view (g) depicts typical bullet-shaped/oval vertebrae (convexity of both the superior and inferior vertebral borders with clear anterior and central beaking) |
Spondyloepiphyseal dysplasia tarda
Spondyloepiphyseal dysplasia tarda (SEDT) is a relatively less frequent osteochondrodysplasia. SEDT is caused by mutations in the TRAPPC2 gene. It is inherited in an X-linked recessive pattern. Affected children demonstrate disproportionately short-trunk short stature with a barrel-shaped chest together with classic radiographic changes in the axial and appendicular skeleton [Figure - 5]. The manifestations are not present at birth but appear later in childhood, typically between ages 6 and 10 years.[2],[3],[19]
![[Figure - 5]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f5.jpg){kind=link}
 |
| Figure 5: (a-d) Spondyloepiphyseal dysplasia tarda. Radiograph of the lumbar spine and hip anteroposterior view (a) in an adolescent patient 1 shows degenerative changes of spine with scoliotic deformity and bilateral hip dysplasia in the form of coxa breva, hip subluxation, shallow, and vertical acetabuli. Follow-up radiographs (b) show significant bilateral arthritic changes. Note the superior-lateral diminution of joint space, subchondral sclerosis. Hand radiograph anteroposterior view of an adolescent patient 2 (c) shows carpal bone amalgamation, sclerosis, and irregularities and the clinical deformity (d) |
Osteogenesis imperfecta
Osteogenesis imperfecta (OI) is a fairly common osteochondrodysplasia. It is the most common genetically determined bone fragility disorder in children. This generalized skeletal connective tissue disorder is characterized chiefly by repeated fractures commonly associated with minimal or no trauma, poor dentition, and blue sclerae. It is caused by mutations in either COL1A1 or COL1A2 and is inherited in an autosomal dominant pattern. Mutations in other recently recognized genes are recessively inherited. These two genes encode the A1 and A2 chains of collagen type I. COL1A1/ 2-related OI has been classified into four types; type I: Classic nondeforming OI with blue sclerae, type II: Perinatally lethal OI, type III: Progressively deforming OI, and type IV: Common variable OI with normal sclerae. A wide array of radioclinical manifestations has been reported ranging from mild susceptibility to fractures up to marked generalized skeletal deformities, disability, and short stature.[2],[3],[19],[20] Differential diagnosis of OI includes disorders that cause generalized skeletal rarefaction as growth hormone deficiency and those with an increased risk of repeated fractures in children as primary hyperparathyroidism and autosomal recessive cutis laxa [Figure - 6].[2],[37] Cutis laxa is an uncommon genetic disorder. The majority of subtypes of cutis laxa syndromes affect connective tissue development through structural gene defects. The skeletal manifestations include spontaneous fractures, joint dislocations, and growth retardation.[38],[39]
![[Figure - 6]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f6.jpg){kind=link}
 |
| Figure 6: (a-h) Osteogenesis imperfecta. Femoral radiograph lateral view (a) of patient 1 demonstrates bilateral asymmetric anterior bowing with healing (solid arrows) and healed (hollow arrows) fractures. Tibial radiograph lateral view (b) demonstrates bilateral anterior tibial bowing “kyphosis”, early healing (solid arrow), progressively healing (hollow arrows), and healed (triangle) fractures. Note the “pencil-like” deformity of both bones, especially the fibulae and the “honeycomb appearance” of the medulla, especially the distal tibias. Pelvis radiograph anteroposterior view patient 2 (c) demonstrates the severe varus deformity of proximal femora and near total healing of subtrochanteric fractures. Note that fractures usually start union on the medial (compressive) surface while delayed union is usually revealed on the lateral (tensile) surface (arrows). The dorsolumbar spine radiograph lateral view of patient 3 (d) shows generalized osteoporosis, biconcave vertebral wedging (hollow arrows), platyspondyly (solid arrow), and various degrees of anterior wedging. Notice that radiographic signs of the spine in osteogenesis imperfecta may closely resemble those of patients receiving growth hormone replacement therapy (e). Note the areas of bone sclerosis intermingled with resorption (e). Humerus radiograph anteroposterior view of a 3-year-old patient 4 (f) demonstrates multiple transverse sclerotic lines (arrows) representing bisphosphonate treatment cycles. Note the elongated thinned out clavicle, ribs, and widened intercostal spaces. The radiographic signs of cutis laxa (g and h) closely resemble osteogenesis imperfecta. Note the united femoral fractures (arrows) in anteroposterior view (g). Upper limb radiograph anteroposterior view (h) shows an apex-lateral angulation of both ulnae, apex-medial angulation of the distal humerus (solid arrows), with dislocated radial heads (hollow arrows), an important differentiating sign from osteogenesis imperfecta |
Mucopolysaccharidosis
Mucopolysaccharidosis (MPS) is a commonly encountered group of osteochondrodysplasias. It is a contiguous gene duplication or deletion syndrome in which multiple genes are involved. All forms of MPS are inherited in an autosomal recessive pattern, with the exception of MPS II; Hunter syndrome which is X-linked. They are caused by disrupted activity of the lysosomal enzymes, which blocks degradation of mucopolysaccharides and leads to accumulation of abnormal byproducts, namely, heparan sulfate, dermatan sulfate, and keratan sulfate. The resulting cellular dysfunction can lead to a variety of skeletal and visceral manifestations. MPS have been subcategorized according to the type of enzyme inadequacy and glycoprotein accumulated as follows: MPS I syndromes (Hurler syndrome, Hurler-Scheie syndrome, or Scheie syndrome), MPS II (Hunter syndrome), MPS III (Sanfilippo syndrome), MPS IV (Morquio syndrome), and MPS VI (Maroteaux-Lamy syndrome) among other less common subtypes. The age of presentation of MPS subtypes is variable. In most types of MPS, the abnormal features are readily distinguishable at birth. Nevertheless, MPS IV (Morquio syndrome) usually presents in children aged 2‘4 years while other types present late in childhood. Significant morbidity can arise from neuro-orthopedic impairment, and the lifespan may be shortened. Hip dysplasia and spinal involvement are prominent musculoskeletal features in patients with MPS.[40],[41],[42],[43] In general, characteristic imaging features are an important aid to the confirmation of MPS diagnosis [Figure - 7]. Employment of imaging features to denote an MPS subtype is challenging. Skeletal MRI may augment the diagnostic role of plain radiographs. MRI may have the potentiality of revealing underlying complications and surgical planning.[14],[15],[44]
![[Figure - 7]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f7.jpg){kind=link}
 |
| Figure 7: (a-e) Mucopolysaccharidosis IV (Morquio syndrome). Pelvis and hip radiograph anteroposterior view (a). Notice the flattened irregular and mottled appearance of the proximal femoral epiphyses, coxa valga (increased neck shaft angle), and wide shallow acetabular cavities. The right hip is dislocated as indicated by a disrupted Shenton's line compared to the relatively undisturbed line on the left hip (white curved lines). The iliac bones are constricted. Pelvis anteroposterior views of another patient with mucopolysaccharidosis demonstrate a bilateral symmetric dislocation of hips with disrupted Shenton's line and femoral heads articulating with false acetabulum (hollow arrow) instead of true acetabulum (solid arrow) (b). The changes demonstrated in the femoral epiphyses of the previous two patients are also typical for Legg–Calvé–Perthes disease (c) and are known as “Perthes-like changes.” Mark that changes exhibited in proximal femoral epiphysis and metaphyses of Perthes disease (c) are typically asymmetrical and the hips exhibit coxa vara with absence of dysplastic changes of the iliac bone. These radiologic signs seen in anteroposterior view of the pelvis provide key clues to distinguish Perthes from mucopolysaccharidosis and other dysplasias. Dorsolumbar spine radiograph lateral view (d) demonstrates kyphosis and various degrees of platyspondyly. Anterior central (solid arrows) and inferior (hollow arrows) beaking is encountered in some vertebrae. Note the expansion of ribs. The hand radiograph anteroposterior view (e) demonstrates pointed proximal ends of second through fifth metacarpals (stars) and metacarpal widening |
Spondylometaphyseal and spondyloepimetaphyseal dysplasia
Spondylometaphyseal dysplasia (SMD) and spondyloepimetaphyseal dysplasia (SEMD) represent a group of osteochondrodysplasias in which the spine and metaphyseal ends of the proximal femora are pivotal features of diagnosis. Heterozygous mutations in the gene encoding the calcium-permeable ion channel TRPV4 on chromosome 12q24 produce SMD. Heterozygous mutations in the type II collagen gene COL2A1 on chromosome 12q13 produce SEMD. Both types are inherited in an autosomal dominant pattern. Short-trunk short stature and narrow thorax are common presenting features.[2],[3] Radiographic features of two children with SMD Kozlowski type and SEMD Strudwick type are depicted [Figure - 8].
![[Figure - 8]](#fig_SaudiOrthopJ_2017_1_2_25_218446_f8.jpg){kind=link}
 |
| Figure 8: (a-e) Spondylometaphyseal dysplasia. Radiographs of pelvis and hip anteroposterior and frog lateral views (a) showing extensive “enchondroma-like” symmetric metaphyseal changes of both femora (arrows). The changes involve areas of bone sclerosis intermingled with areas of resorption. Spine radiograph lateral view (b) demonstrating mild platyspondyly and expansion of anterolateral parts of the ribs with “club-shaped” configuration (arrows). Spondyloepimetaphyseal dysplasia. Radiographs of pelvis and hip anteroposterior view (c) demonstrating extensive metaphyseal broadening of both femora and pelvic bones (star). Note the bilateral femoral epiphyseal involvement and acetabular widening, horizontality and irregularity (hollow arrows). Although changes are asymmetric, both femoral and acetabular components of each hip joint exhibit “congruent incongruity.” These changes are collectively known as “Perthes-like changes” [Figure 7]a and [Figure 7]b. Note the serrated iliac crest (solid arrow). Dorsal spine radiograph lateral view (d) demonstrates extensive platyspondyly (solid arrows), vertebral clefts (hollow arrows), and rib broadening. The leg radiograph anteroposterior views (e) show bilateral genu valgum. Temporary hemiepiphysiodesis was conducted at another hospital aiming for gradual deformity correction. Note the physeal widening and metaphyseal irregularities of proximal tibias (arrows). The presence of clear spinal involvement, pelvic bone changes, and genu valgum in spondyloepimetaphyseal dysplasia are important differentiating signs from Perthes disease |
Conclusion
Skeletal radiography can deliver significant clues to the diagnosis of commonly seen osteochondrodysplasias. The radiologic lesions should be interpreted in light of the clinical features. Adopting a systematized approach to the radiographic assessment of osteochondrodysplasias can optimize the role of diagnostic imaging. Meticulous reporting of the pattern, extent, and severity of radiographic skeletal pathology is an essential part of the diagnostic process.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Authors' contributions
TAE, SME, and NS conceived and designed the study, conducted research, provided, and organized research materials. TAE, RMS, HMS, RG, and SGR collected, analyzed, and interpreted data. TAE wrote initial and final draft of the article. All authors have critically reviewed and approved the final draft, checked the article for plagiarism, and are responsible for the content and similarity index of the manuscript.
| 1. | Chen C, Jiang Y, Xu C, Liu X, Hu L, Xiang Y, et al. Skeleton genetics: A comprehensive database for genes and mutations related to genetic skeletal disorders. Database (Oxford) 2016;2016. pii: baw127. [Google Scholar] |
| 2. | Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015;167A: 2869-92. [Google Scholar] |
| 3. | Krakow D. Skeletal dysplasias. Clin Perinatol 2015;42:301-19, viii. [Google Scholar] |
| 4. | Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med 2010;12:327-41. [Google Scholar] |
| 5. | Ashby E, Eastwood D. Characterization of knee alignment in children with mucopolysaccharidosis types I and II and outcome of treatment with guided growth. J Child Orthop 2015;9:227-33. [Google Scholar] |
| 6. | Karatas AF, Dede O, Rogers K, Ditro CP, Holmes L, Bober M, et al. Growth-sparing spinal instrumentation in skeletal dysplasia. Spine (Phila Pa 1976) 2013;38:E1517-26. [Google Scholar] |
| 7. | EL-Sobky TA, El-Haddad A, Elsobky E, Elsayed SM, Sakr HM. Reversal of skeletal radiographic pathology in a case of malignant infantile osteopetrosis following hematopoietic stem cell transplantation. Egypt J Radiol Nucl Med 2017;48:237-43. [Google Scholar] |
| 8. | Perosky JE, Khoury BM, Jenks TN, Ward FS, Cortright K, Meyer B, et al. Single dose of bisphosphonate preserves gains in bone mass following cessation of sclerostin antibody in Brtl/+ osteogenesis imperfecta model. Bone 2016;93:79-85. [Google Scholar] |
| 9. | Franzone JM, Kruse RW. Intramedullary nailing with supplemental plate and screw fixation of long bones of patients with osteogenesis imperfecta: Operative technique and preliminary results. J Pediatr Orthop B 2016. doi: 10.1097/BPB.0000000000000405. [Google Scholar] |
| 10. | Cho TJ, Lee K, Oh CW, Park MS, Yoo WJ, Choi IH, et al. Locking plate placement with unicortical screw fixation adjunctive to intramedullary rodding in long bones of patients with osteogenesis imperfecta. J Bone Joint Surg Am 2015;97:733-7. [Google Scholar] |
| 11. | Langereis EJ, den Os MM, Breen C, Jones SA, Knaven OC, Mercer J, et al. Progression of hip dysplasia in mucopolysaccharidosis type I hurler after successful hematopoietic stem cell transplantation. J Bone Joint Surg Am 2016;98:386-95. [Google Scholar] |
| 12. | Schmidt M, Breyer S, Löbel U, Yarar S, Stücker R, Ullrich K, et al. Musculoskeletal manifestations in mucopolysaccharidosis type I (Hurler syndrome) following hematopoietic stem cell transplantation. Orphanet J Rare Dis 2016;11:93. [Google Scholar] |
| 13. | Kaissi AA, Hofstaetter J, Weigel G, Grill F, Ganger R, Kircher SG, et al. The constellation of skeletal deformities in a family with mixed types of mucopolysaccharidoses: Case report. Medicine (Baltimore) 2016;95:e4561. [Google Scholar] |
| 14. | Solanki GA, Martin KW, Theroux MC, Lampe C, White KK, Shediac R, et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-brailsford or morquio A syndrome): Presentation, diagnosis and management. J Inherit Metab Dis 2013;36:339-55. [Google Scholar] |
| 15. | Palmucci S, Attinà G, Lanza ML, Belfiore G, Cappello G, Foti PV, et al. Imaging findings of mucopolysaccharidoses: A pictorial review. Insights Imaging 2013;4:443-59. [Google Scholar] |
| 16. | Dhiman N, Albaghdadi A, Zogg CK, Sharma M, Hoover-Fong JE, Ain MC, et al. Factors associated with health-related quality of life (HRQOL) in adults with short stature skeletal dysplasias. Qual Life Res 2017;26:1337-48. [Google Scholar] |
| 17. | Yimgang DP, Brizola E, Shapiro JR. Health outcomes of neonates with osteogenesis imperfecta: A cross-sectional study. J Matern Fetal Neonatal Med 2016;29:3889-93. [Google Scholar] |
| 18. | Hack HA, Walker R, Gardiner P. Anaesthetic implications of the changing management of patients with mucopolysaccharidosis. Anaesth Intensive Care 2016;44:660-8. [Google Scholar] |
| 19. | Panda A, Gamanagatti S, Jana M, Gupta AK. Skeletal dysplasias: A radiographic approach and review of common non-lethal skeletal dysplasias. World J Radiol 2014;6:808-25. [Google Scholar] |
| 20. | Trejo P, Rauch F. Osteogenesis imperfecta in children and adolescents-new developments in diagnosis and treatment. Osteoporos Int 2016;27:3427-37. [Google Scholar] |
| 21. | Cheesman CL, Amirfeyz R, Gargan MF. How to approach a patient with skeletal dysplasia. Orthop Traumatol 2014;28:97-105. [Google Scholar] |
| 22. | Varghese B, Fernandes J. Skeletal dysplasia. Guide to the orthopaedic surgeon. Orthop Trauma 2016;30:500-17. [Google Scholar] |
| 23. | Chung SW, Kang EH, Lee YJ, Ha YJ, Song YW. Three cases of spondyloepiphyseal dysplasia tarda in one Korean family. Yonsei Med J 2016;57:1290-3. [Google Scholar] |
| 24. | Tomatsu S, Yasuda E, Patel P, Ruhnke K, Shimada T, Mackenzie WG, et al. Morquio A syndrome: Diagnosis and current and future therapies. Pediatr Endocrinol Rev 2014;12 Suppl 1:141-51. [Google Scholar] |
| 25. | McKay SD, Al-Omari A, Tomlinson LA, Dormans JP. Review of cervical spine anomalies in genetic syndromes. Spine (Phila Pa 1976) 2012;37:E269-77. [Google Scholar] |
| 26. | Jackson GC, Mittaz-Crettol L, Taylor JA, Mortier GR, Spranger J, Zabel B, et al. Pseudoachondroplasia and multiple epiphyseal dysplasia: A 7-year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution. Hum Mutat 2012;33:144-57. [Google Scholar] |
| 27. | Kim OH, Park H, Seong MW, Cho TJ, Nishimura G, Superti-Furga A, et al. Revisit of multiple epiphyseal dysplasia: Ethnic difference in genotypes and comparison of radiographic features linked to the COMP and MATN3 genes. Am J Med Genet A 2011;155A: 2669-80. [Google Scholar] |
| 28. | EL-Sobky TA, Elsayed SM, El Mikkawy DM. Orthopaedic manifestations of proteus syndrome in a child with literature update. Bone Rep 2015;3:104-8. [Google Scholar] |
| 29. | Alanay Y, Lachman RS. A review of the principles of radiological assessment of skeletal dysplasias. J Clin Res Pediatr Endocrinol 2011;3:163-78. [Google Scholar] |
| 30. | Argente J. Challenges in the management of short stature. Horm Res Paediatr 2016;85:2-10. [Google Scholar] |
| 31. | Gamal R, Elsayed SM, EL-Sobky TA, Elabd HS. Pseudoachondroplasia in a child: the role of anthropometric measurements and skeletal imaging in differential diagnosis. Egypt J Radiol Nucl Med 2017;48:245-50. [Google Scholar] |
| 32. | Sheybani EF, Khanna G, White AJ, Demertzis JL. Imaging of juvenile idiopathic arthritis: A multimodality approach. Radiographics 2013;33:1253-73. [Google Scholar] |
| 33. | Shahnazi M, Khatami A, Shamsian B, Haerizadeh B, Mehrafarin M. Bony Lesions in pediatric acute leukemia: Pictorial essay. Iran J Radiol 2012;9:50-6. [Google Scholar] |
| 34. | Khedr SA, Hassaan MA, Shabanab AA, Gaballah AH, Mokhtar DA. Musculoskeletal manifestations of sickle cell disease, diagnosis with whole body MRI. Egypt J Radiol Nucl Med 2012;43:77-84. [Google Scholar] |
| 35. | Bhuyan BK. Early outcomes of one-stage combined osteotomy in Legg-Calve´-Perthes disease. Indian J Orthop 2016;50:183-94. [Google Scholar] |
| 36. | Tandon A, Bhargava SK, Goel S, Bhatt S. Pseudoachondroplasia: A rare cause of rhizomelic dwarfism. Indian J Orthop 2008;42:477-9. [Google Scholar] |
| 37. | EL-Sobky TA, Ahmad KA, Samir S, EL Mikkawy DM. Primary hyperparathyroidism in a child: The musculoskeletal manifestations of a late presenting rare endocrinopathy. Egypt J Radiol Nucl Med 2016;47:1613-6. [Google Scholar] |
| 38. | Goyal M, Singh A, Kornak U, Kapoor S. The diagnostic dilemma of cutis laxa: A Report of two cases with genotypic dissimilarity. Indian J Dermatol 2015;60:521. [Google Scholar] |
| 39. | Morava E, Guillard M, Lefeber DJ, Wevers RA. Autosomal recessive cutis laxa syndrome revisited. Eur J Hum Genet 2009;17:1099-110. [Google Scholar] |
| 40. | Thawrani DP, Walker K, Polgreen LE, Tolar J, Orchard PJ. Hip dysplasia in patients with Hurler syndrome (mucopolysaccharidosis type 1H). J Pediatr Orthop 2013;33:635-43. [Google Scholar] |
| 41. | Harmatz P, Shediac R. Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment. Front Biosci (Landmark Ed) 2017;22:385-406. [Google Scholar] |
| 42. | Vairo F, Federhen A, Baldo G, Riegel M, Burin M, Leistner-Segal S, et al. Diagnostic and treatment strategies in mucopolysaccharidosis VI. Appl Clin Genet 2015;8:245-55. [Google Scholar] |
| 43. | Kennedy J, Noel J, O'Meara A, Mulhall K, Crushell E, Fogarty E, et al. A long-term retrospective evaluation of functional and radiographic outcomes of pediatric hip surgery in Hurler syndrome. J Pediatr Orthop 2016;36:25-8. [Google Scholar] |
| 44. | White KK, Jester A, Bache CE, Harmatz PR, Shediac R, Thacker MM, et al. Orthopedic management of the extremities in patients with Morquio A syndrome. J Child Orthop 2014;8:295-304. [Google Scholar] |
Fulltext Views
11,246
PDF downloads
2,554




