Translate this page into:
Caffey's disease (Infantile cortical hyperostosis): Case Report, MRI findings, and review of the literature
2 Department of Orthopedics, King Fahad General Hospital, Almadinah Almunawwarah, Saudi Arabia
Corresponding Author:
Abdularahman H Refai
Department of Surgery, Orthopedic Surgery Division, National Guard Health System, Prince Mohammed Bin Abdulaziz Hospital, National Guard, P.O. Box 40740 Almadinah Almunawwarah 41511
Saudi Arabia
drrefaii@gmail.com
| How to cite this article: Refai AH, Taha WS, Almahdi H, Arabi H. Caffey's disease (Infantile cortical hyperostosis): Case Report, MRI findings, and review of the literature. J Musculoskelet Surg Res 2018;2:173-176 |
Abstract
Caffey's disease or infantile cortical hyperostosis is a rare condition that affects infants and is mostly a self-limiting condition. It presents as an acute inflammatory process of the periosteum and the surrounding soft tissues. Affected infants present with irritability and fever. In many cases, the diagnosis gets delayed because of the rarity of the condition and the fact that it mimics a wide range of diseases that are more common and more serious including acute osteomyelitis, bone tumors, scurvy, hypervitaminosis A, infantile malignant osteopetrosis, and child abuse. This case is reported to get the awareness of the physicians about the disease, its existence in our population, presentation, and important differential diagnoses and management and to show the important role of magnetic resonance imaging in the management.Introduction
Caffey's disease or infantile cortical hyperostosis can present in two forms, a (classical) mild infantile form and the severe prenatal form. The classical form which was described by Caffey's and Silverman is the mild infantile form. They reported it as a disease entity in 1945 and it was described as a rare disease with no preference in gender or race.[1] The classic form of the disease is more common than the prenatal form. Caffey's disease is a rare, self-limiting disease characterized by a subperiosteal reaction and new bone formation involving the diaphysis of the mandible and long bones. It usually affects the infants before 5 months of age and subsides before the age of 2 years.[2] The exact prevalence is unknown as it resolves spontaneously, which leads to underdiagnosis. The incidence is fluctuating and may be influenced by environmental effects.[3] Its exact etiology is still unknown.[4] There are many proposed causes; among them are infections, immunological defects, and genetic abnormalities. Most cases are sporadic, but a few familial cases with both autosomal dominant and autosomal recessive have been reported.[3],[5] The autosomal dominant inheritance was discovered in three unrelated families (gene COLIAL, 17q21), which encodes alfa-1 chain of type I collagen that has raised some doubts about whether some cases are a type of collagenopathy, as osteogenesis imperfecta.[6],[7] One case has been reported in an infant with cyanotic heart disease following prolonged treatment with prostaglandin E1 (PGE1) for ductal patency maintenance.[8],[9] Patients can present with a painful swelling in the affected area and fever.[2] There is no specific laboratory test to diagnose Caffey's disease, but it can present with leukocytosis, increased erythrocyte sedimentation rate (ESR), and alkaline phosphatase (ALP), indicating an inflammatory response. In radiographs, it appears as a new bone formation, or periosteal reaction mimicking an infectious process, which should be ruled out along with other differential diagnoses as tumors, child abuse, chronic hypervitaminosis A, scurvy, malignant infantile osteopetrosis,[10] and prolonged PGE1 infusion.[8],[9] The disease shows a good response to nonsteroidal anti-inflammatory drugs (NSAIDs). Recurrence and adverse outcomes are very rare.[11] Patients can present with a triad of symptoms, signs, and radiographic picture. The usual presentation is irritability and swelling of the overlying soft tissue that usually precedes the cortical thickening of the underlying bones associated with low-grade fever and anorexia. The swelling is painful with a wood-like induration but with no redness or warmth. There are usually no other signs and symptoms. The most commonly involved site is the mandibular bone, followed by scapula, clavicle, ribs, and long bones. Some isolated cases of Erb's palsy and facial nerve palsy have been reported in the literature.[12],[13] In cases of severe pain, the pain can result in pseudoparalysis, and some rare clinical findings can be present as nasal obstruction, proptosis, and dysphagia.[14],[15],[16] The severe prenatal onset form is characterized by extensive hyperostotic bone involvement, which may lead to the wrong diagnosis of the lethal form of osteogenesis imperfecta.[4],[11],[17],[18] This case report is written to present the first case of Caffey's disease reported in our region and to highlight its existence, presentation, and important differential diagnoses to avoid unnecessary invasive investigations and treatments in the affected infants and to show the important role of magnetic resonance imaging (MRI) in excluding many of the important differential diagnoses.
Case Report
An 8-month-old boy presented to the emergency room (ER), with pain and swelling in the right forearm for 2 weeks and undocumented fever, with a history of one ER visit for right mandible swelling 3 weeks before the onset of the right forearm pain and swelling. The patient was otherwise healthy; his birth history was uneventful, with normal development, and his vaccination was up-to-date.
On examination, the patient was irritable, with stable vital signs except for low-grade fever, with tenderness and mild swelling of the right forearm; no redness or hotness was appreciated. There were no discharging sinuses or lymphadenopathy. He had a full range of motion of the shoulder, elbow, and wrist. The mandible has improved and was not tender. The rest of the examination was unremarkable [Figure - 1].
![[Figure - 1]](#fig_SaudiOrthopJ_2018_2_4_173_243700_f1.jpg){kind=link}
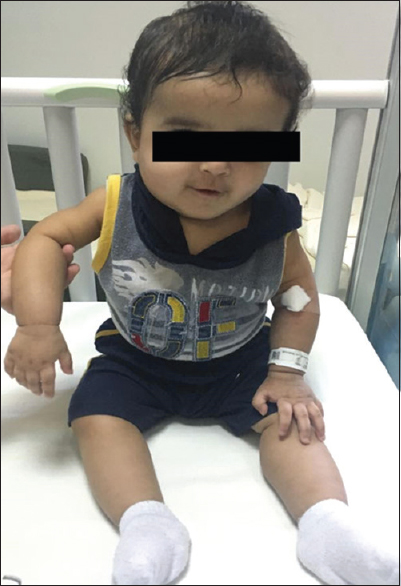 |
| Figure 1: Picture of the patient showing the right forearm and mandible swelling |
His initial ER investigations showed white blood cell count of 14.6 × 103/μL, with 37.60% neutrophils, hemoglobin of 10.3 g/dL, platelet count of 743 × 109/L, C-reactive protein 141.2, and ESR 105 mm/h. The liver function tests, ALP, and coagulation profile were within normal limits; also, blood culture was done and showed no growth.
The radiographs showed periosteal reaction along the whole length of the right radius with a suspicion of a linear nondisplaced fracture at the distal metaphysis of the right radius. However, there was no history of trauma; so, we considered it as a radiological artifact, which did not show in follow-up radiographs. A minimal periosteal reaction was also noted around the proximal part of the right ulna [Figure - 2]a and [Figure - 2]b.
![[Figure - 2]](#fig_SaudiOrthopJ_2018_2_4_173_243700_f2.jpg){kind=link}
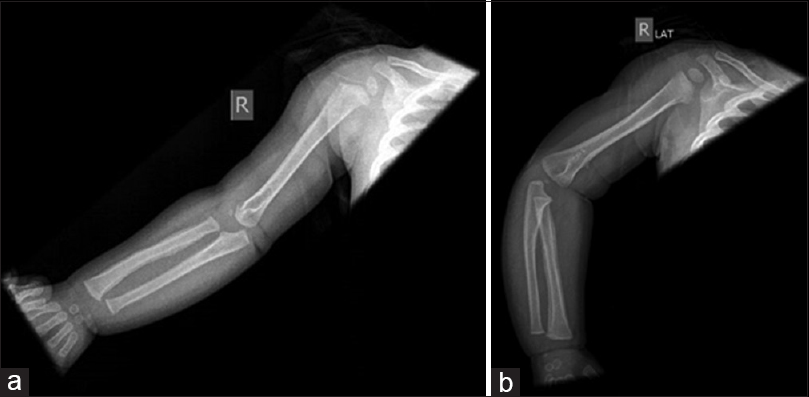 |
| Figure 2: (a and b) Anteroposterior and lateral radiographs of the right upper limb (at initial presentation) with the arrows pointing to the areas of periosteal thickening of the whole radius |
The patient was admitted as a possible case of osteomyelitis and after consultation with general pediatric and infectious disease services. An initial working diagnosis of osteomyelitis was made, and he was started on intravenous cefazolin as an empirical treatment, and ibuprofen was given orally for pain. The patient's condition did not improve on the antibiotic given. Skeletal survey was requested and in addition to the finding mentioned previously, it also revealed evidence of linear, smooth, and thick periosteal reaction around the borders of the mandible with increased thickness [Figure - 2]a, [Figure - 2]b and [Figure - 3]. The MRI showed homogenous diffuse circumferential mild thickening and abnormal signal intensity with pathological enhancement of the periosteum and a periosteal reaction involving the whole scanned length of both right radius and ulna. No visible involvement of the right wrist joint was noted. Minimal scattered fluid signal intensity was seen within the dorsal soft tissues of the forearm; however, no drainable-enhancing collection was appreciated. There was no apparent bone destruction appreciated [Figure - 4].
![[Figure - 3]](#fig_SaudiOrthopJ_2018_2_4_173_243700_f3.jpg){kind=link}
![[Figure - 4]](#fig_SaudiOrthopJ_2018_2_4_173_243700_f4.jpg){kind=link}
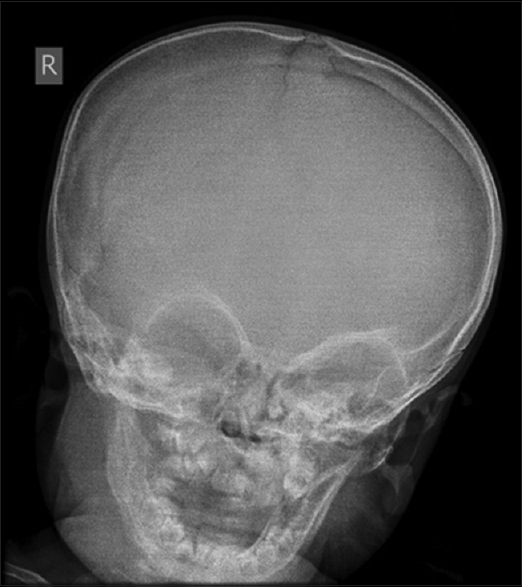 |
| Figure 3: Radiograph of the skull (at the initial presentation) with the arrows pointing to the areas of periosteal thickening. Note: both mandibles are showing periosteal reaction, more on the right side |
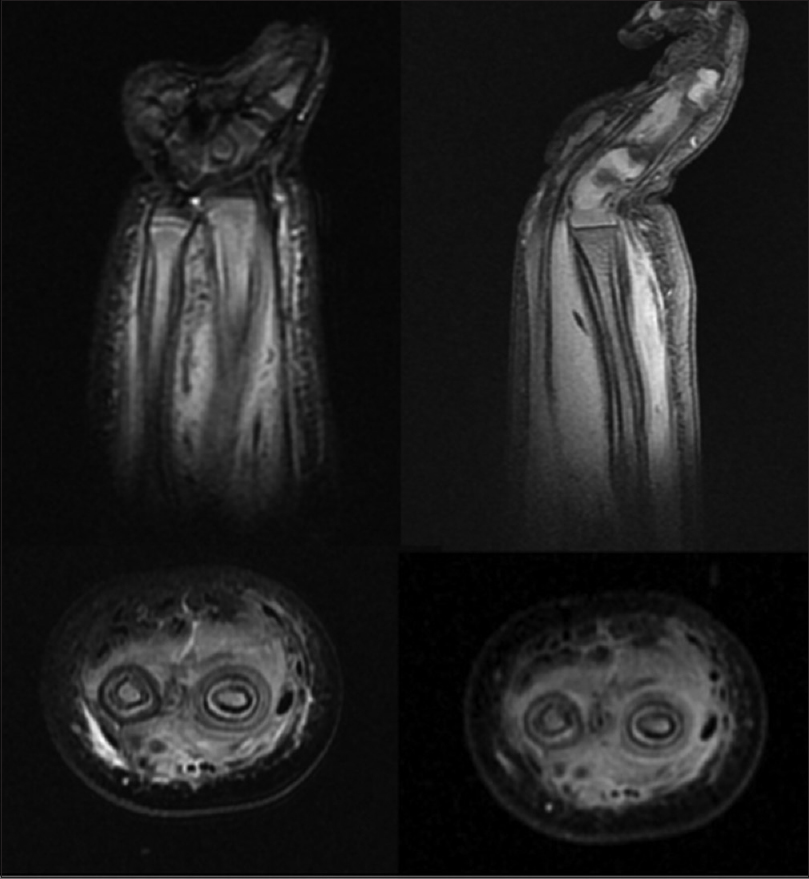 |
| Figure 4: Magnetic Resonance Imaging showing homogenous diffuse circumferential mild thickening and abnormal signal intensity with pathological enhancement of the periosteum and a periosteal reaction involving the whole scanned length of both right radius and ulna |
Hence, the diagnosis was changed from osteomyelitis to Caffey's disease based on the findings and the discussion between the treating specialties. The patient was discharged home after he became afebrile and less irritable. The patient was discharged on acetaminophen when needed.
The patient was seen at the outpatient department after 1 week for follow-up when his temperature was stable, he became less irritable, and also the forearm swelling decreased dramatically with very little tenderness. We followed the patient in the clinic after 6 weeks, 3 months, 6 months, and 1 year with skeletal survey radiographs to monitor the previously detected lesions and to look for new lesions. In each visit, he was completely free of symptoms with no more tenderness of his forearm and a full range of motion of all joints. Fortunately, no more lesions occurred, and the forearm and mandible bone hypertrophy showed great improvement without causing any deformity apart from an increased cortical thickness [Figure - 5].
![[Figure - 5]](#fig_SaudiOrthopJ_2018_2_4_173_243700_f5.jpg){kind=link}
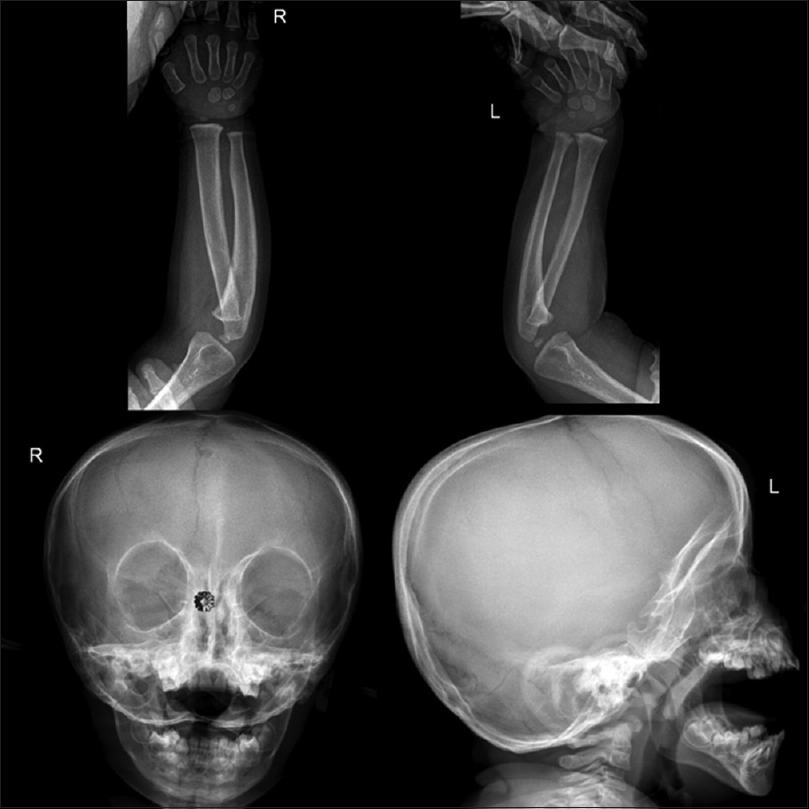 |
| Figure 5: Follow-up radiographs of the forearm and skull (6 months after discharge) showing complete resorption of the periosteal reaction and replacement by normal bone, but with an increase in the diameter of the radius |
Discussion
Our current case had none of the uncommon symptoms and signs of Caffey's disease such as nasal obstruction, proptosis, and dysphagia. In the literature, the reported laboratory tests include anemia and elevated ESR and in some patients raised ALP, thrombocytosis, and elevated immunoglobulin levels.[19],[20] Our patient had anemia, elevated ESR, and thrombocytosis, but the ALP was not elevated. Unfortunately, immunoglobulin test was not done.
Although radiography is not diagnostic in Caffey's disease, it is the most valuable diagnostic study to exclude the important differential diagnoses. A characteristic feature is the new cortical bone formation beneath the regions of the soft-tissue swelling seen in the radiograph of the affected site. The MRI showing more detailed information about the soft-tissue reactions and edema can help exclude like in our case bone infection and tumors. As there is no specific laboratory test for the diagnosis of Caffey's disease, the exclusion of osteomyelitis is necessary. Bone tumor such as Ewing sarcoma which is the second most common malignant bone tumor in children is another important differential diagnosis of Caffey's disease. It presents with localized swelling and pain and appears on radiography as a permeative bone destruction. Ewing sarcoma needs to be excluded with certainty as early management is critical to have a good prognosis.[21]
The other differential diagnoses are child abuse, chronic hypervitaminosis A, scurvy, and prolonged PGE1 infusion.[8],[9],[18],[22] Infantile malignant osteopetrosis should be considered as a differential (with its three types infantile malignant autosomal recessive osteopetrosis, intermediate autosomal recessive osteopetrosis, and autosomal dominant osteopetrosis) as if it is untreated, it has a fatal outcome. The condition is commonly diagnosed in infancy with symptoms of significant hematologic abnormalities with bone marrow failure, hepatosplenomegaly, macrocephaly with frontal bossing, and bone fractures.[10]
Caffey's disease is mostly a self-limiting disease and resolves within 6–12 months and may not need any intervention. However, NSAID could be used in severe symptomatic cases.[22] In cases of poor response to NSAID, steroids can be used. In our case, we used ibuprofen during the admission, and the patient was discharged on acetaminophen, and the outcome was satisfactory. The bone lesions can recur at any time at the previous sites or different sites, and these relapses can happen even several years later. The clinical course with remissions and relapses can be unpredictable.[6],[14] Awareness of the existence of this rare condition and its typical clinicoradiological profile will protect the patient from getting subjected to multiple unnecessary investigations and treatments.[10],[23]
Conclusion
Caffey's disease is an uncommon disease presenting with inconsistent clinical manifestations simulating osteomyelitis and needs a high index of suspicion and an awareness of the existence of this condition. A detailed history, good clinical examination, basic laboratory studies, and plain radiographs are sufficient enough to make a diagnosis in most cases, and MRI can help excluding the important differential diagnoses.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Internal institution support.
Conflicts of interest
There is no conflict of interest.
Author's contributions
AR, WT, HM and HA contributed to the design and implementation of the research, collection of the data to the analysis of the results and to the writing of the manuscript. All authors read and approved the final manuscript. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
| 1. | Caffey J, Silverman W. Infantile cortical hyperostosis, preliminary report of a new syndrome. Am J Roentgenol Radiat Ther 1945;54:1-16. [Google Scholar] |
| 2. | Gensure RC, Mäkitie O, Barclay C, Chan C, Depalma SR, Bastepe M, et al. Anovel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest 2005;115:1250-7. [Google Scholar] |
| 3. | Hall C. Caffey Disease. Orphanet Encyclopedia; February, 2005. Available from: http://www.orpha.net. [Last accessed on 2018 May]. [Google Scholar] |
| 4. | Restrepo S, Sánchez AM, Palacios E. Infantile cortical hyperostosis of the mandible. Ear Nose Throat J 2004;83:454-5. [Google Scholar] |
| 5. | Bernstein RM, Zaleske DJ. Familial aspects of Caffey's disease. Am J Orthop (Belle Mead NJ) 1995;24:777-81. [Google Scholar] |
| 6. | Glorieux FH. Caffey disease: An unlikely collagenopathy. J Clin Invest 2005;115:1142-4. [Google Scholar] |
| 7. | Caffey Disease a Type 1 Collagenopathy, Current GGH. Vol. 24; September, 2005. Available from: http://www.GGHjournal.com. [Last accessed on 2018 May]. [Google Scholar] |
| 8. | de Almeida JF, Kimura H, Hercowitz LH, Korkes H, Troster EJ. Cortical hyperostosis secondary to prolonged use of prostaglandin E1. Clinics (Sao Paulo) 2007;62:363-6. [Google Scholar] |
| 9. | Nadroo AM, Shringari S, Garg M, al-Sowailem AM. Prostaglandin induced cortical hyperostosis in neonates with cyanotic heart disease. J Perinat Med 2000;28:447-52. [Google Scholar] |
| 10. | Mazzolari E, Forino C, Razza A, Porta F, Villa A, Notarangelo LD, et al. Asingle-center experience in 20 patients with infantile malignant osteopetrosis. Am J Hematol 2009;84:473-9. [Google Scholar] |
| 11. | Saul RA, Lee WH, Stevenson RE. Caffey's disease revisited. Further evidence for autosomal dominant inheritance with incomplete penetrance. Am J Dis Child 1982;136:55-60. [Google Scholar] |
| 12. | Challapalli M, Cunningham DG, Varnado SC. Infantile cortical hyperostosis and facial nerve palsy. Int J Pediatr Otorhinolaryngol 1998;43:175-8. [Google Scholar] |
| 13. | Holtzman D. Infantile cortical hyperostosis of the scapula presenting as an ipsilateral Erb's palsy. J Pediatr 1972;81:785-8. [Google Scholar] |
| 14. | Schweiger S, Chaoui R, Tennstedt C, Lehmann K, Mundlos S, Tinschert S, et al. Antenatal onset of cortical hyperostosis (Caffey disease): Case report and review. Am J Med Genet A 2003;120A: 547-52. [Google Scholar] |
| 15. | Sheppard JJ, Pressman H. Dysphagia in infantile cortical hyperostosis (Caffey's disease): A case study. Dev Med Child Neurol 1988;30:111-4. [Google Scholar] |
| 16. | Fauré C, Beyssac JM, Montagne JP. Predominant of exclusive orbital and facial involvement in infantile cortical hyperostosis (de toni-Caffey's disease). Report of four cases and a review of the literature. Pediatr Radiol 1977;6:103-6. [Google Scholar] |
| 17. | Mohammed AL. Caffey Silverman disease: Case report and literature review. Kuwait Med J 2006;38:49-52. [Google Scholar] |
| 18. | Almada Rodriguez Hugo D. Non Accidental Injuries in Children-Common Pit Falls. Oman Med J 2010;25:134-6. [Google Scholar] |
| 19. | Temperley IJ, Douglas SJ, Rees JP. Raised immunoglobulin levels and thrombocytosis in infantile cortical hyperostosis. Arch Dis Child 1972;47:982-3. [Google Scholar] |
| 20. | Kumar TS, Scott JX, Mathew LG. Caffey disease with raised immunoglobulin levels and thrombocytosis. Indian J Pediatr 2008;75:181-2. [Google Scholar] |
| 21. | Hawkins DS, Brennan B, Bölling T, Davidson DJ, Dirksen U, DuBois S, et al. Ewing sarcoma. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia: Lippincott Williams and Wilkins; 2015. p. 854-75. [Google Scholar] |
| 22. | Dutta S, Jain N, Bhattacharya A, Mukhopadhyaya K. Infantile cortical hyperostosis. Indian Paediatr 2002;39:1057. [Google Scholar] |
| 23. | ALBagshi MH, ALZoayed HI. Infantile cortical hyperostosis – A report of Saudi family. Sudan J Paediatr 2015;15:61-4. [Google Scholar] |
Fulltext Views
4,441
PDF downloads
615




