Translate this page into:
Genetics of developmental dysplasia of the hip: Recent progress and future perspectives
2 Department of Orthopedic, College of Medicine, Taibah University, Almadinah Almunawwarah, Kingdom of Saudi Arabia
Corresponding Author:
Sulman Basit
Center for Genetics and Inherited Diseases, Taibah University, Almadinah Almunawwarah
Kingdom of Saudi Arabia
sbasit.phd@gmail.com
| How to cite this article: Hashmi JA, Basit S, Khoshhal KI. Genetics of developmental dysplasia of the hip: Recent progress and future perspectives. J Musculoskelet Surg Res 2019;3:245-253 |
Abstract
Developmental dysplasia of the hip (DDH) is the most common congenital orthopedic disorder in infants. DDH is a heterogeneous disorder, and the exact pathophysiology is incompletely understood; however, several environmental as well as genetic factors have been identified as an underlying player in its pathogenesis. Involvement of genetic factors in the pathogenesis is evident from the fact that DDH occurrence has been observed in families with multiple affected individuals. Here, we reviewed the current literature on DDH, specifically concentrating on the genetic aspects of isolated (nonsyndromic) form of DDH. It is observed that genetic association studies, as well as linkage study designs, have been used to identify the extent of involvement of genetic factors in DDH pathogenesis. Variants in genes involved in joint development and chondrogenesis including HOXD9, ASPN, HOXB9, TGF-Beta 1, PAPPA2, DKK1, and GDF5 genes have been identified as associated with DDH through genetic association studies. Moreover, mutations in CX3CR1 and TENM3 have been identified using linkage analysis and exome sequencing. Although various approaches including association studies, linkage analysis, and targeted sequencing have been used to detect genetic factors underlying DDH, structural genetic variants underlying DDH have not been explored. Therefore, we propose that copy number variation analysis and interactome studies (interaction analysis using gene and protein data using molecular interaction search tools) can help in identifying new DDH-associated genes.Introduction
Developmental dysplasia of the hip (DDH: MIM 142700) is a congenital orthopedic malformation of the hip joint leading to a distorted femoral head socket. Globally, DDH exceeds all other congenital orthopedic disorders in terms of disease prevalence.[1],[2],[3] Anatomically, DDH is hallmarked by aberrant acetabular and/or femoral growth, which is implicated in a typical dysplasia encompassing hip joint dislocation or subluxation leading to impaired joint functioning. The impaired articular surface apposition may lead to early arthritis.[4],[5] Phenotypic spectrum of DDH manifests variability, which is linked to the degree of aberration found in the femoral head and acetabulum. DDH is a heterogeneous disorder where several environmental as well as genetic factors play their role in its pathogenesis.[1],[2],[3],[4],[5],[6] Among environmental factors, several pregnancy-related physiological conditions such as lack of amniotic fluid (oligohydramnios), breech presentation whereby buttocks and/or feet of the newborn baby comes first during delivery, delivering for the first time (primiparity), and increased weight of the newly born baby have been associated with the DDH pathogenesis.[3],[7],[8],[9],[10] The mere fact that more than one member of a single family are affected by DDH [11],[12],[13],[14],[15] depicts a pivotal role of genetic factors underlying DDH pathogenesis.[16],[17],[18],[19] A substantial increase in disease risk of DDH has been reported in Asian siblings of affected families, with double impact in case of female siblings compared to male siblings.[20],[21] DDH has been divided into DDH1 (MIM 142700) and DDH2 (MIM 615612) based on genetic heterogeneity. The inheritance pattern in DDH1 is multifactorial and the disease phenotype has been mapped on chromosome 13q22,[13] whereas DDH2 shows autosomal dominant inheritance and has been mapped on chromosome 3p22.2.[4] Clinical heterogeneity of DDH can be seen in its syndromic forms, where it is shown up with other clinical disorders such as cardiac and renal malformations and club foot,[22] though mostly it is manifested as an isolated entity without any other additional disorders. This review focuses on the genetic aspects of isolated form of DDH. The disease incidence rate of DDH varies in different parts of the world, ranging normally 1.5–20 cases out of 1000 live births,[23] with even higher rates of DDH incidence for certain Mediterranean countries along with Italy and Japan.[24],[25] This variation in incidence is because of different practicing parameters such as the time at which the DDH is evaluated clinically and the use of different diagnostic methods.[23] An incidence rate of 3.17–3.50 for every 1000 live births has been documented for the Middle East, based on certain hospital-based studies.[26],[27],[28] This figure may rise considerably provided a detailed population-based study at the national level is conducted in the Middle Eastern countries especially in the Kingdom of Saudi Arabia, where genetic disorders are relatively more prevalent due to a typical tribal culture and common consanguineous marriages.[29]
In this article, we have reviewed genetic studies on DDH that are available online in PubMed. Keywords including “developmental dysplasia of the hip” in combination with “genetics,” “gene,” “mutation,” “congenital,” and “inherited” were used to search for the relevant literature. We found that a variety of genetic approaches have been used to identify the genetic defects underlying DDH based on the type of samples available and the pattern of inheritance of the disease. We first discuss the approaches used to delineate the genetics of DDH, followed by brief details of the genetic variants reported in DDH cases and families.
A. Genetic Association Studies
Detection of association of genetic variants in a specific gene or genomic region with a given phenotype is usually carried out using the technique of genetic association. In this approach, genetic markers are genotyped in a group of DNA samples from patients (cases) and normal individuals (controls). Nowadays, single-nucleotide polymorphism (SNP) markers are extensively used for genotyping. Screening of SNPs determines the association of a phenotype with a particular area of a chromosome. This association is helpful in exposing the underlying genes for a particular trait.[30] The International Human HapMap Project has generated human genome haplotypes, which are used in genotyping SNPs throughout the genome. This approach is termed as genome-wide association studies (GWASs).
In GWAS, the whole genome is screened using SNPs; therefore, a good number of patients as well as control samples from the same population are required.[31],[32] In the context of DDH, recruiting enough patients for GWAS always remains a challenge. This led to comparatively less efficiency of GWAS in detecting DDH-specific susceptibility genetic variants. However, few case–control studies have led to the identification of genetic variants underlying DDH phenotype in several populations. Susceptible variants in genes involved in joint development and chondrogenesis have been associated with DDH. These include variants in HOXD9, ASPN, HOXB9, TGF-Beta 1, PAPPA2, DKK1, and GDF5 genes.[10],[33],[34],[35],[36],[37],[38],[39]
B. Genetic Linkage Studies
Studying the inheritance pattern of a phenotype in a family and detection of genetic markers closely segregating with the disease phenotype has been used extensively to identify the underlying genetic variants in several diseases. This approach has identified mutations in several genes as a causative factor of the genetic diseases. Earlier, identification of chromosomal parts segregating with a disease phenotype within a family was accomplished using microsatellite markers. Nowadays, SNP markers are genotyped throughout the genome to detect linkage. This approach is termed as genome-wide linkage analysis (GWLA). GWLA approach is not very effective in complex genetic disorders as compared to single-gene disorders.[40],[41] In DDH, owing to complex inheritance fashion and incomplete penetrance of the potentially pathogenic variants, GWLA has not successfully mapped chromosomal regions except in few studies.[3],[4],[15] Moreover, a variable spectrum of DDH phenotype is also observed in different members of a single family manifesting its phenotypic heterogeneity.[42] The chromosomal regions identified in linkage to DDH using GWLA approach include regions on chromosome 3p22.2-p22.1, 4q35, 13q22, 16p, and 17q21.32.[4],[13],[15],[43]
C. Structural Genomic Variations
Structural genomic aberrations, which lead to a change in the diploid pattern of the genome, in the form of deletions or duplications, are known as copy number variants (CNVs). Moreover, chromosomal rearrangements typically larger than 1 kb are also classified as structural genomic variations. Different genome analytic approaches such as whole-genome SNP genotyping, array comparative genomic hybridization, and high-throughput DNA sequencing are generally used to detect CNVs. Currently, high-resolution microarrays having probes for both the SNPs and CNVs are used as a gold standard to identify the CNVs.[44] Disease-causing CNVs manifest disease phenotypes by either distorting the coding region of the gene or increasing the number of the already existing gene or by giving rise to a new gene from the fusion of two genes. At present, above 6,359,956 CNVs are enlisted in the database of genomic variants (2016). Likewise, 22,358 disease-causing CNVs (gross insertions and deletions) are documented in the Human Gene Mutation Database (2019). These CNVs have been implicated in several genetic disorders such as schizophrenia, bipolar disorders, and autism.[45],[46],[47],[48] As far as DDH is concerned, both sporadic and familial, no study has been conducted to identify CNVs in DDH. Any such study may come up with novel results.
D. DNA Sequencing
Sequencing coding part of the human genome or even the complete human genome can be used to detect mutations underlying DDH. Sequencing of complete protein-coding regions (exons) of the genome is termed as whole-exome sequencing (WES). WES covers almost 3% of the human genome. Another DNA sequencing approach, called whole-genome sequencing (WGS), determines the order of all the nucleotides in an individual's DNA and can determine variations in any part of the genome. With the advent of high-throughput next-generation sequencing technique, the dream of sequencing the entire coding regions of the genome (WES) and WGS in a matter of days with accuracy and reliability has changed into reality. The very first implication of WES in the identification of mutations underlying a genetic disorder was first reported in 2009.[49] There onward, this powerful technique has successfully been implicated in finding the underlying genetic variants in a variety of genetic disorders ranging from skeletal abnormalities, neurological malformations, skin disorders, and retinal dystrophies.[50],[51],[52],[53] Feldman et al. in 2013[4] recruited a large family with multiple individuals affected with DDH. WES of the family led to the identification of a genetic variant in a chemokine receptor gene (CX3CR1). Previously, this nonsynonymous variant (rs3732378) was thought to be an SNP. The authors have shown that this variant is indeed a pathogenic variant using different pathogenicity prediction software such as PolyPhen-2 and SIFT. Based on the presence of phenotypic variability among the affected members of a DDH family, the authors have also suggested the role of a modifier variant that might have been involved in disease heterogeneity. This indicates the need for further investigations and validations of the variant (rs3732378) in CX3CR1 gene. Recently, the same group has reported a mutation in teneurin 3 (TENM3) in a large family with multiple DDH-affected individuals.[54]
Single and Small Genomic Variations in Developmental Dysplasia of the Hip
Using the above-mentioned approaches, many genetic variants were found affiliated with DDH or as an underlying genetic defect in DDH. Variants in genes such as CX3CR1, TENM3, PAPPA2, COL2A1, HOXD9, GDF-5, and TGFB1 have been identified as increasing the susceptibility of having DDH.[55]
Chemokine-CX3C motif-receptor 1
Chemokine-CX3C motif-receptor 1 (CX3CR1) is a member of a group of about 45 proteins of human chemokine family that play a significant role in human health and diseases development. Chemokines are small protein molecules that express in response to injury or infection and bind to and subsequently activate chemokine receptors. This led to the leukocyte adhesion to the vessel wall, leukocyte trafficking, changes in the morphology, and chemotaxis to the site of injury or infection.[56] Moreover, activation of chemokine receptor via chemokine binding also plays a role in different biological processes such as extracellular matrix remodeling and tumor metastasis besides differentiation and activation.[57],[58],[59],[60] Furthermore, HIV and malarial parasite use chemokine receptors to invade the host cells.[61],[62]
Using an approach of linkage analysis and WES, Feldman et al.[25] identified a variant in CX3CR1 as an underlying genetic defect in a large family from Utah. DNA samples from 71 members of a family were collected, and DNA from four severely affected members was exome sequenced. A previously reported population polymorphism (rs3732378) in CX3CR1 was identified as a pathogenic variant. Moreover, few sporadic DDH cases were screened, and the same variant was detected.[21],[25] In continuation of their previous work, Feldman et al. generated a CK3CR1 knocked down mice and compared the resultant phenotype with the wild-type mice. Computerized tomography (CT) was used to evaluate the hips of both the knocked down and wild-type mice at the age of 5 and 8 weeks, respectively. An inclined treadmill was used to evaluate the gait of 8-week-old mice. The authors showed that CX3CR1 ablation affects acetabular morphology and gait.[63]
In a case–control study, Li et al. found that variants in CX3CR1 increase the susceptibility to DDH. The assumption is based on genotyping data from 689 DDH patients, in which two CX3CR1 variants (rs3732378 and rs3732379) were genotyped.[21]
Teneurin transmembrane protein 3
TENM3 encodes a transmembrane protein, which belongs to the tenascin superfamily. Members of this family play a variety of functions. TENM3 predominantly functions in the development of the visual system.[64],[65] TENM3 mutations have been reported to cause different disease conditions including motor developmental delay, ocular coloboma, microphthalmia, and intellectual disability.[66],[67],[68],[69],[70],[71],[72],[73],[74],[75],[76],[77],[78] Although developing nervous system harbors high levels of TENM3, TENM3 mRNA can also be found in prechondrogenic mesenchymal cells. Therefore, it could be involved in the initial phase of differentiation of chondrogenic cells.[69] Recently, the mutation in TENM3 has been found in a family segregating DDH.[54] A mouse model was generated for TENM3 mutation depicting late left glenoid fossa and acetabular development. Moreover, it was found that the bone marrow cells from TENM3 mutant mouse overexpress MMP13 with or without BMP2 stimulation. It is known that higher levels of MMP13 lead to cartilage degeneration.[70] Based on the above observations, the authors hypothesized that mutated TENM3 may slow chondrogenesis.[54]
WNT1-inducible signaling pathway protein 3
WNT1-inducible signaling pathway (WISP) protein subfamily comes under the umbrella of connective tissue growth factor family. WISP3 encodes a member of WISP family. Genes that belong to this family are involved in regulating cell growth and differentiation.[71],[72] Mutations in the WISP3 gene have been known to cause an autosomal recessive form of progressive pseudorheumatoid dysplasia (PDD). PDD is a skeletal disorder characterized by damaged hip joint as a result of continuous degeneration and loss of articular cartilage.[71],[73]
Recently, a case–control study, including 386 DDH patients and 558 healthy individuals, identified an SNP (rs69306665) upstream of WISP3 gene in association with DDH.[1]
Ubiquinol-cytochrome c reductase complex assembly factor 1
Ubiquinol-cytochrome c reductase complex assembly factor 1 (UQCC1) is reported to be expressed in developing cartilaginous cells called chondrocytes, which are responsible for the secretion of the matrix by the cartilage.[74] This gene has previously been reported as a candidate gene for phenotypes such as height, testicular germ cell tumor, and spine bone size.[75],[76],[77] Keeping in view the importance of UQCC in the chondrification process, an associated role of UQCC with DDH had been suggested. The results of GWASs revealed 12 variants in UQCC1 gene linked with DDH. Following GWAS, a case–control study was conducted to evaluate the association of UQCC gene in DDH manifestation. The results confirmed the association of UQCC1 variant (rs6060373) with DDH phenotype in the Han Chinese population.[30]
Asporin
A cartilaginous protein called Asporin (ASPN) is encoded by the gene ASPN. ASPN is involved in the regulation of cartilage development process, chondrogenesis.[78],[79],[80] ASPN is reported to bind with bone morphogenetic protein 2 (BMP2) and subsequently block the downstream BMP/Smad signaling pathway.[81],[82] BMP2 is an important growth factor of transforming growth factor β1 (TGF-β1) family and plays a significant role in the proliferation and differentiation of osteoblast and perichondrial cells.[83],[84] The gene comprises a repeat region of aspartic acid, which is linked with skeletal anomalies, owing to its polymorphism.[79]
Skeletal abnormalities such as lumbar disc malformation, rheumatoid arthritis, and osteoarthritis of the hip region are affiliated with allelic polymorphism of the ASPN gene.[85],[86],[87] A recent study identified loss of copy number variations in ASPN gene on chromosome 9 at 9q22.31 and found that the CNVs in the region are associated with severe acetabular dysplasia.[87] Moreover, a case–control study established the association of ASPN polymorphism and DDH.[34] The authors showed that polymorphism in ASPN actually affects TGF-β1 to cause DDH.
Transforming growth factor-beta 1
TGF-β1-encoded protein was found to be involved in different developmental processes such as growth and proliferation of cells followed by their differentiation. The protein is also considered to play a regulatory role for certain other growth factors.[88],[89],[90],[91] To evaluate the role of TGF-β1 in DDH, a carefully designed case–control study was conducted. The cases included were osteoarthritis patients secondary to DDH and the controls were osteoarthritis patients without any DDH manifestation. This investigation identified a potential interaction between TGF-β1 and interleukin 6 (IL6). Moreover, this study found an association of variants in both TGF-β1 and IL6 with DDH.[6]
Growth differentiation factor 5
Proteins involved in the formation of bone skeleton are coded by GDF5 and lie under the umbrella of a superfamily of proteins known as TGF-β. Binding different receptors in signal transduction pathways, these proteins regulate expression of the genes.[92] GDF5 is involved in the morphogenesis of different cell types including neural and bone formation-related cell types beside teeth and fat cell types.[93] Any change in the sequence of this gene will lead to skeletal-related anomalies.[94],[95],[96],[97],[98],[99],[100],[101],[102],[103],[104],[105],[106]
As far as the hip is concerned, GDF5 has been implicated in the development of an embryonic limb/skeletal system, in particular development of articular cavities and cartilage.[93],[106],[107],[108],[109] GDF5 is an important player involved in hip development and joint formation.[108] Several GWAS established the link between common SNPs spanning a 130 kb interval containing GDF5 and osteoarthritis.[110],[111],[112] Moreover, various reproducible studies showed a strong association of GDF5 with DDH and hip dislocation.[33],[113],[114],[115]
Pregnancy-associated plasma protein-A2
Protein generated by PAPPA2 interacts with binding protein 5 of insulin-like growth factor (IGF) and is considered to regulate the IGF.[116],[117] Pregnancy-associated plasma protein-A2 (PAPPA2) is identified to have a major regulatory role in the growth process, so the effect of PAPPA2 on bone shape and size has been suggested.[118] Deletion of PAPPA2 in mice leads to shorter femur length.[119] In humans, an association between DDH and a PAPPA2 SNP has been reported.[36] Others have replicated the results of previous studies.[120]
Homeobox genes
Homeobox (HOX) gene-encoded transcription factors occupy a key position in the development of the vertebrate skeleton. This group contains 39 genes and are mapped to four gene loci HOXA-D.[121] HOX genes regulate target genes by binding the targeted DNA region using its homeodomain.[122] For instance, anomalies related to lower limbs are related to 5' truncation of HOXC genes.[123] In the following part, we discuss only those genes having known association with DDH.
HOXD genes encompass nine genes, namely HOXD1, 3, 4, 8, 9, 10, 11, 12, and 13, which are closely located at chromosome 2q24.1-q33.1. These genes have their role known in skeletal system development, particularly in the development of a limb.[124] By creating different knockdown mutant models for HOXD genes in mice and chicken led to the finding that any perturbance in the expression pattern of HOXD genes will lead to change in shape and size of the targeted skeletal part.[125],[126] The positioning of the hip joint in proximal limb part suggest a coincidence between hip area and area of expression of 9th pairs of HOX gene. Keeping in view these facts, a hypothesis was made regarding a possible role of HOXD in DDH manifestation. To verify this hypothesis, a genetic association study was designed to assess the association between SNPs of HOXD9 gene and female Han Chinese population with DDH symptoms. The findings of the study revealed the connection between HOXD9 SNPs (rs711819) and Han Chinese population with DDH. The studies also reported HOXD9 as a novel disease candidate gene for DDH.[37] Follow-up and functional studies are required to clearly elucidate the exact molecular mechanism through which this part of the genome is associated pathogenically with DDH.
HOXB9 is a member of the Abd-B HOX family and has implications in cellular developmental processes such as proliferation and subsequent differentiation. As far as DDH pathogenicity is concerned, HOXB9 gene appears with contradictory results. In one of the studies where genetic linkage analysis was done for a Chinese DDH family, DDH was shown to be linked to chromosome 17 at position 17q21 where HOXB9 gene is located.[127] However, these findings were contradicted in a case–control study in a European population where no association was developed between SNPs of HOXB9 and DDH.[128] Another study where whole-genome screening established linkage to a specific location on chromosome 17 where HOXB9 gene is also present.[15] Looking up at these contradictory reports, a case–control study on the Han Chinese population was conducted to evaluate the association between HOXB9 and DDH manifestation. Two tag SNPs (rs8844 and rs2303486) were used in the study. This study established an association of HOXB9 SNP (rs2303486) and DDH.[38]
T-box 4
Transcription factors that regulate different developmental mechanisms are encoded by T-box 4 (TBX4), one of the T-box genes.[129] Expression of Tbx4 in growing hindlimb in chicken and mouse models indicates a connection between this gene and regulation and specification of limb development.[130],[131] Microduplications involving TBX4 are associated with clubfoot in humans.[132] Moreover, mutations in TBX4 cause a small patella syndrome, a skeletal dysplasia, in which cartilage and bone growth processes are affected. As a result, individuals with small patella syndrome have malformations of the pelvis and feet.[133] Likewise, limb tissue-specific Tbx4 mutant mice showed skeletal abnormalities such as hypoplastic pelvis, femurs, and fibula.[134] Keeping in mind the significant role played by Tbx4 in skeletal development, especially hindlimb development, it has been assumed that Tbx4 SNPs might cause susceptibility to DDH phenotype. To evaluate this assumption, all SNPs of Tbx4 were evaluated and two SNPs (rs3744438 and rs3744448) were found to be associated with DDH.[135]
Developmental dysplasia of the hip-associated genes
Some DDH-associated genes such as ASPN, GDF5, TGFB1, and HOX are interrelated functionally. However, genes such as TENM3, CX3CR1, and PAPPA2 are not related to each other and with other DDH genes. Similarly, HSPG2 and ATP2B4 gene mutations are known in familial DDH; however, they are not functionally interacting with other DDH known genes.[2] Interaction analysis using gene and protein data using molecular interaction search tools such as MIST [136] and GeneMANIA [137],[138] was carried out to identify functional interaction between DDH-associated genes and to detect new candidate gene. For instance, using molecular interactome analysis, we identified several members of keratin-associated (KRTAP) genes that directly interact with DDH-associated gene. KRTAP members identified in this analysis include KRTAP1-1, KRTAP1-5, KRTAP2-3, KRTAP3-1, KRTAP3-2, KRTAP4-2, KRTAP4-11, KRTAP4-12, KRTAP5-7, KRTAP5-9, KRTAP6-2, KRTAP10-8, KRTAP10-11, KRTAP12-2, KRTAP17-1, and KRTAP19-5. The KRTAP proteins form a matrix of keratin intermediate filaments which contribute to the structure of hair fibers. KRTAP family members appear to have unique, family-specific amino- and carboxyl-terminal regions. Their association with disease conditions has not been described yet. Mutations in these genes might contribute to the development of DDH. We found that most of the DDH genes interact with each other either directly through protein–protein interaction or indirectly through transcription factors. Interactome analysis using gene and protein data identified several important genes as well [Figure - 1].
![[Figure - 1]](#fig_SaudiOrthopJ_2019_3_3_245_264146_f1.jpg){kind=link}
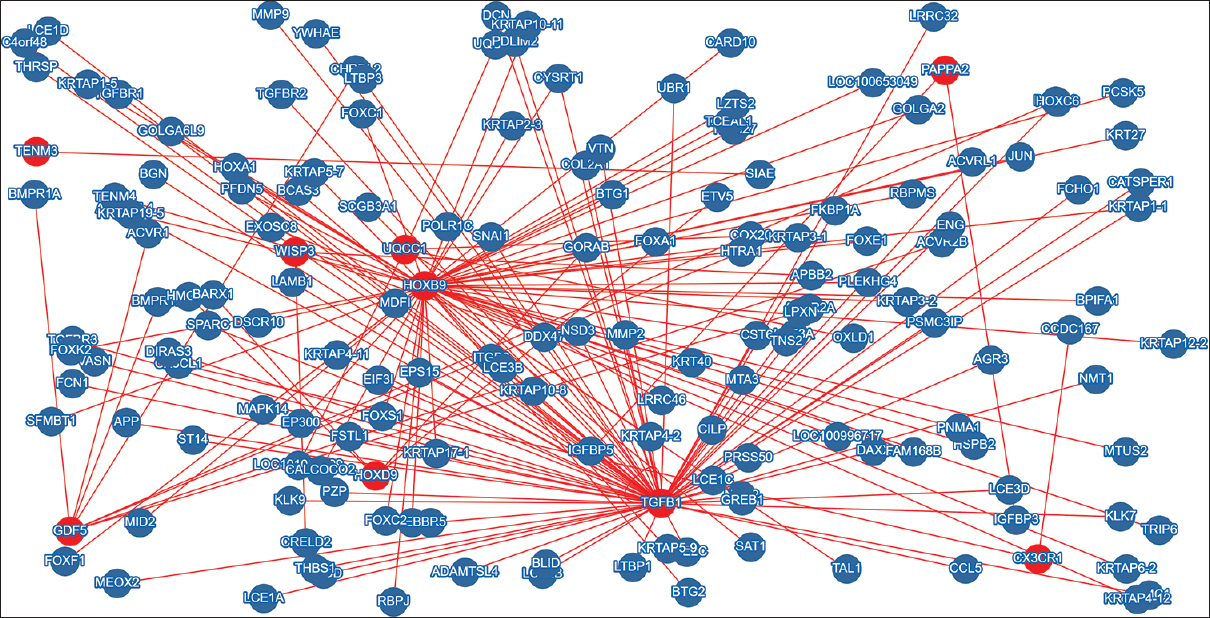 |
| Figure 1: Output of molecular interaction search tool. Developmental dysplasia of the hip-associated genes were used as a training set to identify molecular interactions between developmental dysplasia of the hip genes. Other potential candidate genes have also been identified |
Conclusion
Development dysplasia of the hip is clinically and genetically a heterogeneous disorder. It encompasses a broad range of disorders including minor acetabular dysplasia to irreducible hip dislocation. The pathophysiology of the DDH is incompletely understood. Mild hip subluxations and dislocation escape the early diagnosis due to lack of optimal timing for clinical examination, imaging, and appropriate use of imaging.
Genetic studies have helped in identifying the molecular markers for DDH. Association studies and linkage analysis succeeded in identifying several candidate genes, such as PAPPA2, COL2A1, HOXD9, GDF-5, TGFB1, CX3CR1, and TENM3 as DDH-associated genes. It is believed that these genes play a significant role in the pathogenesis of DDH. A genetic screening program can be established for the screening for mild forms of DDH. Genetic screening program also has the potential to detect the DDH earlier in life.
Some of the DDH genes discussed in this review are part of the same signaling pathways, or they interact with each other through protein–protein interaction. For instance, ASPN, GDF5, TGFB1, and HOX genes are interrelated functionally. However, some genes such as TENM3, CX3CR1, and PAPPA2 are not related to each other and with other DDH genes. Similarly, HSPG2 and ATP2B4 gene mutations are known in familial DDH; however, they are not functionally interacting with other DDH known genes. Using DDH known genes, molecular interaction search tools have been used to identify other interacting genes in DDH-related signaling pathways. Interactome analysis has the potential to identify new potential candidate genes for studies aiming at identifying genetic variants underlying DDH.
Identification of candidate genes would pave the way for early detection of DDH. Timely detection helps in preventing further disability including osteoarthritis and movement impairment and improves the psychological health and quality of life in affected children.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Authors' contribution
KIK conceived the idea. JAH and SB performed literature survey and wrote the initial draft. SB carried out molecular interaction studies. KIK edited the initial draft and provided guidelines for improvement of the initial draft. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
| 1. | Zhang J, Yan M, Zhang Y, Yang H, Sun Y. Association analysis on polymorphisms in WISP3 gene and developmental dysplasia of the hip in Han Chinese population: A case-control study. Gene 2018;664:192-5. [Google Scholar] |
| 2. | Basit S, Albalawi AM, Alharby E, Khoshhal KI. Exome sequencing identified rare variants in genes HSPG2 and ATP2B4 in a family segregating developmental dysplasia of the hip. BMC Med Genet 2017;18:34. [Google Scholar] |
| 3. | Basit S, Alharby E, Albalawi AM, Khoshhal KI. Whole genome SNP genotyping in a family segregating developmental dysplasia of the hip detected runs of homozygosity on chromosomes 15q13.3 and 19p13.2. Congenit Anom (Kyoto) 2018;58:56-61. [Google Scholar] |
| 4. | Feldman GJ, Parvizi J, Levenstien M, Scott K, Erickson JA, Fortina P, et al. Developmental dysplasia of the hip: Linkage mapping and whole exome sequencing identify a shared variant in CX3CR1 in all affected members of a large multigeneration family. J Bone Miner Res 2013;28:2540-9. [Google Scholar] |
| 5. | Jacobsen S, Sonne-Holm S, Søballe K, Gebuhr P, Lund B. Hip dysplasia and osteoarthrosis: A survey of 4151 subjects from the osteoarthrosis substudy of the Copenhagen city heart study. Acta Orthop 2005;76:149-58. [Google Scholar] |
| 6. | Čengić T, Trkulja V, Pavelić SK, Ratkaj I, Markova-Car E, Mikolaučić M, et al. Association of TGFB1 29C/T and IL6 -572G/C polymorphisms with developmental hip dysplasia: A case-control study in adults with severe osteoarthritis. Int Orthop 2015;39:793-8. [Google Scholar] |
| 7. | de Hundt M, Vlemmix F, Bais JM, Hutton EK, de Groot CJ, Mol BW, et al. Risk factors for developmental dysplasia of the hip: A meta-analysis. Eur J Obstet Gynecol Reprod Biol 2012;165:8-17. [Google Scholar] |
| 8. | Ortiz-Neira CL, Paolucci EO, Donnon T. A meta-analysis of common risk factors associated with the diagnosis of developmental dysplasia of the hip in newborns. Eur J Radiol 2012;81:e344-51. [Google Scholar] |
| 9. | Patel H; Canadian Task Force on Preventive Health Care. Preventive health care, 2001 update: Screening and management of developmental dysplasia of the hip in newborns. CMAJ 2001;164:1669-77. [Google Scholar] |
| 10. | Shi D, Dai J, Ikegawa S, Jiang Q. Genetic study on developmental dysplasia of the hip. Eur J Clin Invest 2012;42:1121-5. [Google Scholar] |
| 11. | Cilliers HJ, Beighton P. Beukes familial hip dysplasia: An autosomal dominant entity. Am J Med Genet 1990;36:386-90. [Google Scholar] |
| 12. | Tönnis D, Heinecke A. Acetabular and femoral anteversion: Relationship with osteoarthritis of the hip. J Bone Joint Surg Am 1999;81:1747-70. [Google Scholar] |
| 13. | Mabuchi A, Nakamura S, Takatori Y, Ikegawa S. Familial osteoarthritis of the hip joint associated with acetabular dysplasia maps to chromosome 13q. Am J Hum Genet 2006;79:163-8. [Google Scholar] |
| 14. | Ceylaner G, Ceylaner S, Ustünkan F, Inan M. Autosomal dominant inheritance of congenital dislocation of the hip in 16 members of a family. Acta Orthop Traumatol Turc 2008;42:289-91. [Google Scholar] |
| 15. | Feldman G, Dalsey C, Fertala K, Azimi D, Fortina P, Devoto M, et al. The Otto Aufranc award: Identification of a 4 Mb region on chromosome 17q21 linked to developmental dysplasia of the hip in one 18-member, multigeneration family. Clin Orthop Relat Res 2010;468:337-44. [Google Scholar] |
| 16. | Woolf CM, Koehn JH, Coleman SS. Congenital hip disease in Utah: The influence of genetic and nongenetic factors. Am J Hum Genet 1968;20:430-9. [Google Scholar] |
| 17. | Czeizel A, Szentpétery J, Tusnády G, Vizkelety T. Two family studies on congenital dislocation of the hip after early orthopaedic screening Hungary. J Med Genet 1975;12:125-30. [Google Scholar] |
| 18. | Kramer AA, Berg K, Nance WE. Familial aggregation of congenital dislocation of the hip in a Norwegian population. J Clin Epidemiol 1988;41:91-6. [Google Scholar] |
| 19. | Wilkinson JA. Etiologic factors in congenital displacement of the hip and myelodysplasia. Clin Orthop Relat Res 1992;281:75-83. [Google Scholar] |
| 20. | Li L, Sun K, Zhang L, Zhao Q, Cheng X, Dang Y. Heritability and sibling recurrent risk of developmental dysplasia of the hip in Chinese population. Eur J Clin Invest 2013;43:589-94. [Google Scholar] |
| 21. | Li L, Wang X, Zhao Q, Wang E, Wang L, Cheng J, et al. CX3CR1 polymorphisms associated with an increased risk of developmental dysplasia of the hip in human. J Orthop Res 2017;35:377-80. [Google Scholar] |
| 22. | Karapinar L, Sürenkök F, Oztürk H, Us MR, Yurdakul L. The importance of predicted risk factors in developmental hip dysplasia: An ultrasonographic screening program. Acta Orthop Traumatol Turc 2002;36:106-10. [Google Scholar] |
| 23. | Bialik V, Bialik GM, Blazer S, Sujov P, Wiener F, Berant M. Developmental dysplasia of the hip: A new approach to incidence. Pediatrics 1999;103:93-9. [Google Scholar] |
| 24. | Rubini M, Cavallaro A, Calzolari E, Bighetti G, Sollazzo V. Exclusion of COL2A1 and VDR as developmental dysplasia of the hip genes. Clin Orthop Relat Res 2008;466:878-83. [Google Scholar] |
| 25. | Feldman GJ, Parvizi J, Sawan H, Erickson JA, Peters CL. Linkage mapping and whole exome sequencing identify a shared variant in CX3CR1 in a large multi-generation family. J Arthroplasty 2014;29:238-41. [Google Scholar] |
| 26. | Mirdad T. Incidence and pattern of congenital dislocation of the hip in Aseer region of Saudi Arabia. West Afr J Med 2002;21:218-22. [Google Scholar] |
| 27. | Kremli MK, Alshahid AH, Khoshhal KI, Zamzam MM. The pattern of developmental dysplasia of the hip. Saudi Med J 2003;24:1118-20. [Google Scholar] |
| 28. | Moosa NK, Kumar PT, Mahmoodi SM. Incidence of developmental dysplasia of the hip in Dubai. Saudi Med J 2009;30:952-5. [Google Scholar] |
| 29. | Sun Y, Wang C, Hao Z, Dai J, Chen D, Xu Z, et al. Acommon variant of ubiquinol-cytochrome c reductase complex is associated with DDH. PLoS One 2015;10:e0120212. [Google Scholar] |
| 30. | Hannan M, Basit S, Almontashiri NA, Khoshhal KI. The need for population-based studies to estimate the rate of consanguinity in Almadinah Almunawwarah. J Taibah Univ Med Sci 2015;10:509-11. [Google Scholar] |
| 31. | Chatterjee N, Chen YH, Luo S, Carroll RJ. Analysis of case-control association studies: SNPs, imputation and haplotypes. Stat Sci 2009;24:489-502. [Google Scholar] |
| 32. | Laird NM, Lange C. Family-based designs in the age of large-scale gene-association studies. Nat Rev Genet 2006;7:385-94. [Google Scholar] |
| 33. | Dai J, Shi D, Zhu P, Qin J, Ni H, Xu Y, et al. Association of a single nucleotide polymorphism in growth differentiate factor 5 with congenital dysplasia of the hip: A case-control study. Arthritis Res Ther 2008;10:R126. [Google Scholar] |
| 34. | Shi D, Dai J, Zhu P, Qin J, Zhu L, Zhu H, et al. Association of the D repeat polymorphism in the ASPN gene with developmental dysplasia of the hip: A case-control study in Han Chinese. Arthritis Res Ther 2011;13:R27. [Google Scholar] |
| 35. | Kolundžić R, Trkulja V, Mikolaučić M, Kolundžić MJ, Pavelić SK, Pavelić K. Association of interleukin-6 and transforming growth factor-β1 gene polymorphisms with developmental hip dysplasia and severe adult hip osteoarthritis: A preliminary study. Cytokine 2011;54:125-8. [Google Scholar] |
| 36. | Jia J, Li L, Zhao Q, Zhang L, Ru J, Liu X, et al. Association of a single nucleotide polymorphism in pregnancy-associated plasma protein-A2 with developmental dysplasia of the hip: A case-control study. Osteoarthritis Cartilage 2012;20:60-3. [Google Scholar] |
| 37. | Tian W, Zhao L, Wang J, Suo P, Wang J, Cheng L, et al. Association analysis between HOXD9 genes and the development of developmental dysplasia of the hip in Chinese female Han population. BMC Musculoskelet Disord 2012;13:59. [Google Scholar] |
| 38. | Hao Z, Dai J, Shi D, Xu Z, Chen D, Zhao B, et al. Association of a single nucleotide polymorphism in HOXB9 with developmental dysplasia of the hip: A case-control study. J Orthop Res 2014;32:179-82. [Google Scholar] |
| 39. | Liu S, Tian W, Wang J, Cheng L, Jia J, Ma X. Two single-nucleotide polymorphisms in the DKK1 gene are associated with developmental dysplasia of the hip in the Chinese Han female population. Genet Test Mol Biomarkers 2014;18:557-61. [Google Scholar] |
| 40. | Strachan T, Read A. Human Molecular Genetics. 4th ed. New York: Garland Science; 2011. p. 467-95. [Google Scholar] |
| 41. | Altmüller J, Palmer LJ, Fischer G, Scherb H, Wjst M. Genomewide scans of complex human diseases: True linkage is hard to find. Am J Hum Genet 2001;69:936-50. [Google Scholar] |
| 42. | Feldman GJ, Peters CL, Erickson JA, Hozack BA, Jaraha R, Parvizi J. Variable expression and incomplete penetrance of developmental dysplasia of the hip: Clinical challenge in a 71-member multigeneration family. J Arthroplasty 2012;27:527-32. [Google Scholar] |
| 43. | Loughlin J, Mustafa Z, Irven C, Smith A, Carr AJ, Sykes B, et al. Stratification analysis of an osteoarthritis genome screen-suggestive linkage to chromosomes 4, 6, and 16. Am J Hum Genet 1999;65:1795-8. [Google Scholar] |
| 44. | Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559-75. [Google Scholar] |
| 45. | Grayton HM, Fernandes C, Rujescu D, Collier DA. Copy number variations in neurodevelopmental disorders. Prog Neurobiol 2012;99:81-91. [Google Scholar] |
| 46. | Boone PM, Yuan B, Campbell IM, Scull JC, Withers MA, Baggett BC, et al. The alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. Am J Hum Genet 2014;95:143-61. [Google Scholar] |
| 47. | Chaudhry A, Noor A, Degagne B, Baker K, Bok LA, Brady AF, et al. Phenotypic spectrum associated with PTCHD1 deletions and truncating mutations includes intellectual disability and autism spectrum disorder. Clin Genet 2015;88:224-33. [Google Scholar] |
| 48. | Georgieva L, Rees E, Moran JL, Chambert KD, Milanova V, Craddock N, et al. De novo CNVs in bipolar affective disorder and schizophrenia. Hum Mol Genet 2014;23:6677-83. [Google Scholar] |
| 49. | Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009;461:272-6. [Google Scholar] |
| 50. | Çalışkan M, Chong JX, Uricchio L, Anderson R, Chen P, Sougnez C, et al. Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum Mol Genet 2011;20:1285-9. [Google Scholar] |
| 51. | Takeichi T, Nanda A, Liu L, Salam A, Campbell P, Fong K, et al. Impact of next generation sequencing on diagnostics in a genetic skin disease clinic. Exp Dermatol 2013;22:825-31. [Google Scholar] |
| 52. | Rainger J, Pehlivan D, Johansson S, Bengani H, Sanchez-Pulido L, Williamson KA, et al. Monoallelic and biallelic mutations in MAB21L2 cause a spectrum of major eye malformations. Am J Hum Genet 2014;94:915-23. [Google Scholar] |
| 53. | Li D, Weber DR, Deardorff MA, Hakonarson H, Levine MA. Exome sequencing reveals a nonsense mutation in MMP13 as a new cause of autosomal recessive metaphyseal anadysplasia. Eur J Hum Genet 2015;23:264-6. [Google Scholar] |
| 54. | Feldman G, Kappes D, Mookerjee-Basu J, Freeman T, Fertala A, Parvizi J. Novel mutation in teneurin 3 found to co-segregate in all affecteds in a multi-generation family with developmental dysplasia of the hip. J Orthop Res 2019;37:171-80. [Google Scholar] |
| 55. | Zamborsky R, Kokavec M, Harsanyi S, Attia D, Danisovic L. Developmental dysplasia of hip: Perspectives in genetic screening. Med Sci (Basel) 2019;7. pii: E59. [Google Scholar] |
| 56. | Moser B, Wolf M, Walz A, Loetscher P. Chemokines: Multiple levels of leukocyte migration control. Trends Immunol 2004;25:75-84. [Google Scholar] |
| 57. | Helmke A, Nordlohne J, Balzer MS, Dong L, Rong S, Hiss M, et al. CX3CL1-CX3CR1 interaction mediates macrophage-mesothelial cross talk and promotes peritoneal fibrosis. Kidney Int 2019;95:1405-17. [Google Scholar] |
| 58. | Dimberg A. Chemokines in angiogenesis. Curr Top Microbiol Immunol 2010;341:59-80. [Google Scholar] |
| 59. | Speyer CL, Ward PA. Role of endothelial chemokines and their receptors during inflammation. J Invest Surg 2011;24:18-27. [Google Scholar] |
| 60. | Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev 2006;25:357-71. [Google Scholar] |
| 61. | Choe H, Moore MJ, Owens CM, Wright PL, Vasilieva N, Li W, et al. Sulphated tyrosines mediate association of chemokines and Plasmodium vivax duffy binding protein with the duffy antigen/receptor for chemokines (DARC). Mol Microbiol 2005;55:1413-22. [Google Scholar] |
| 62. | Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, et al. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996;272:1955-8. [Google Scholar] |
| 63. | Feldman G, Offemaria A, Sawan H, Parvizi J, Freeman TA. A murine model for developmental dysplasia of the hip: Ablation of CX3CR1 affects acetabular morphology and gait. J Transl Med 2017;15:233. [Google Scholar] |
| 64. | Antinucci P, Nikolaou N, Meyer MP, Hindges R. Teneurin-3 specifies morphological and functional connectivity of retinal ganglion cells in the vertebrate visual system. Cell Rep 2013;5:582-92. [Google Scholar] |
| 65. | Merlin S, Horng S, Marotte LR, Sur M, Sawatari A, Leamey CA. Deletion of ten-m3 induces the formation of eye dominance domains in mouse visual cortex. Cereb Cortex 2013;23:763-74. [Google Scholar] |
| 66. | Singh B, Srivastava P, Phadke SR. Sequence variations in TENM3 gene causing eye anomalies with intellectual disability: Expanding the phenotypic spectrum. Eur J Med Genet 2019;62:61-4. [Google Scholar] |
| 67. | Stephen J, Nampoothiri S, Kuppa S, Yesodharan D, Radhakrishnan N, Gahl WA, et al. Novel truncating mutation in TENM3 in siblings with motor developmental delay, ocular coloboma, oval cornea, without microphthalmia. Am J Med Genet A 2018;176:2930-3. [Google Scholar] |
| 68. | Chassaing N, Ragge N, Plaisancié J, Patat O, Geneviève D, Rivier F, et al. Confirmation of TENM3 involvement in autosomal recessive colobomatous microphthalmia. Am J Med Genet A 2016;170:1895-8. [Google Scholar] |
| 69. | Murakami T, Fukunaga T, Takeshita N, Hiratsuka K, Abiko Y, Yamashiro T, et al. Expression of ten-m/Odz3 in the fibrous layer of mandibular condylar cartilage during postnatal growth in mice. J Anat 2010;217:236-44. [Google Scholar] |
| 70. | Ning B, Sun J, Yuan Y, Yao J, Wang P, Ma R. Early articular cartilage degeneration in a developmental dislocation of the hip model results from activation of β-catenin. Int J Clin Exp Pathol 2014;7:1369-78. [Google Scholar] |
| 71. | Nakamura Y, Weidinger G, Liang JO, Aquilina-Beck A, Tamai K, Moon RT, et al. The CCN family member Wisp3, mutant in progressive pseudorheumatoid dysplasia, modulates BMP and Wnt signaling. J Clin Invest 2007;117:3075-86. [Google Scholar] |
| 72. | Pal A, Huang W, Li X, Toy KA, Nikolovska-Coleska Z, Kleer CG. CCN6 modulates BMP signaling via the Smad-independent TAK1/p38 pathway, acting to suppress metastasis of breast cancer. Cancer Res 2012;72:4818-28. [Google Scholar] |
| 73. | Hurvitz JR, Suwairi WM, Van Hul W, El-Shanti H, Superti-Furga A, Roudier J, et al. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat Genet 1999;23:94-8. [Google Scholar] |
| 74. | Imabayashi H, Mori T, Gojo S, Kiyono T, Sugiyama T, Irie R, et al. Redifferentiation of dedifferentiated chondrocytes and chondrogenesis of human bone marrow stromal cells via chondrosphere formation with expression profiling by large-scale cDNA analysis. Exp Cell Res 2003;288:35-50. [Google Scholar] |
| 75. | Deng FY, Dong SS, Xu XH, Liu YJ, Liu YZ, Shen H, et al. Genome-wide association study identified UQCC locus for spine bone size in humans. Bone 2013;53:129-33. [Google Scholar] |
| 76. | Cook MB, Chia VM, Berndt SI, Graubard BI, Chanock SJ, Rubertone MV, et al. Genetic contributions to the association between adult height and testicular germ cell tumors. Int J Epidemiol 2011;40:731-9. [Google Scholar] |
| 77. | Sanna S, Jackson AU, Nagaraja R, Willer CJ, Chen WM, Bonnycastle LL, et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet 2008;40:198-203. [Google Scholar] |
| 78. | Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. A novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem 2001;276:12201-11. [Google Scholar] |
| 79. | Kizawa H, Kou I, Iida A, Sudo A, Miyamoto Y, Fukuda A, et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet 2005;37:138-44. [Google Scholar] |
| 80. | Nakajima M, Kizawa H, Saitoh M, Kou I, Miyazono K, Ikegawa S. Mechanisms for asporin function and regulation in articular cartilage. J Biol Chem 2007;282:32185-92. [Google Scholar] |
| 81. | Yamada S, Tomoeda M, Ozawa Y, Yoneda S, Terashima Y, Ikezawa K, et al. PLAP-1/asporin, a novel negative regulator of periodontal ligament mineralization. J Biol Chem 2007;282:23070-80. [Google Scholar] |
| 82. | Tomoeda M, Yamada S, Shirai H, Ozawa Y, Yanagita M, Murakami S. PLAP-1/asporin inhibits activation of BMP receptor via its leucine-rich repeat motif. Biochem Biophys Res Commun 2008;371:191-6. [Google Scholar] |
| 83. | Samee M, Kasugai S, Kondo H, Ohya K, Shimokawa H, Kuroda S. Bone morphogenetic protein-2 (BMP-2) and vascular endothelial growth factor (VEGF) transfection to human periosteal cells enhances osteoblast differentiation and bone formation. J Pharmacol Sci 2008;108:18-31. [Google Scholar] |
| 84. | Lecanda F, Avioli LV, Cheng SL. Regulation of bone matrix protein expression and induction of differentiation of human osteoblasts and human bone marrow stromal cells by bone morphogenetic protein-2. J Cell Biochem 1997;67:386-96. [Google Scholar] |
| 85. | Song YQ, Cheung KM, Ho DW, Poon SC, Chiba K, Kawaguchi Y, et al. Association of the asporin D14 allele with lumbar-disc degeneration in Asians. Am J Hum Genet 2008;82:744-7. [Google Scholar] |
| 86. | Torres B, Orozco G, García-Lozano JR, Oliver J, Fernández O, González-Gay MA, et al. Asporin repeat polymorphism in rheumatoid arthritis. Ann Rheum Dis 2007;66:118-20. [Google Scholar] |
| 87. | Sekimoto T, Ishii M, Emi M, Kurogi S, Funamoto T, Yonezawa Y, et al. Copy number loss in the region of the ASPN gene in patients with acetabular dysplasia: ASPN CNV in acetabular dysplasia. Bone Joint Res 2017;6:439-45. [Google Scholar] |
| 88. | Sandell LJ. Etiology of osteoarthritis: Genetics and synovial joint development. Nat Rev Rheumatol 2012;8:77-89. [Google Scholar] |
| 89. | Malemud CJ. Anticytokine therapy for osteoarthritis: Evidence to date. Drugs Aging 2010;27:95-115. [Google Scholar] |
| 90. | Roelen BA, Dijke P. Controlling mesenchymal stem cell differentiation by TGFBeta family members. J Orthop Sci 2003;8:740-8. [Google Scholar] |
| 91. | Lieskovska J, Guo D, Derman E. IL-6-overexpression brings about growth impairment potentially through a GH receptor defect. Growth Horm IGF Res 2002;12:388-98. [Google Scholar] |
| 92. | Liu J, Saito K, Maruya Y, Nakamura T, Yamada A, Fukumoto E, et al. Mutant GDF5 enhances ameloblast differentiation via accelerated BMP2-induced Smad1/5/8 phosphorylation. Sci Rep 2016;6:23670. [Google Scholar] |
| 93. | Kiapour AM, Cao J, Young M, Capellini TD. The role of Gdf5 regulatory regions in development of hip morphology. PLoS One 2018;13:e0202785. [Google Scholar] |
| 94. | Thomas JT, Kilpatrick MW, Lin K, Erlacher L, Lembessis P, Costa T, et al. Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet 1997;17:58-64. [Google Scholar] |
| 95. | Ullah A, Umair M, Hussain S, Jan A, Ahmad W. Sequence variants in GDF5 and TRPS1 underlie brachydactyly and tricho-rhino-phalangeal syndrome type III. Pediatr Int 2018;60:304-6. [Google Scholar] |
| 96. | Khan S, Mudassir M, Khan N, Marwat A. Brachdactyly instigated as a result of mutation in GDF5 and NOG genes in Pakistani population. Pak J Med Sci 2018;34:82-7. [Google Scholar] |
| 97. | Umair M, Rafique A, Ullah A, Ahmad F, Ali RH, Nasir A, et al. Novel homozygous sequence variants in the GDF5 gene underlie acromesomelic dysplasia type-grebe in consanguineous families. Congenit Anom (Kyoto) 2017;57:45-51. [Google Scholar] |
| 98. | Basit S, Naqvi SK, Wasif N, Ali G, Ansar M, Ahmad W. A novel insertion mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene underlies Grebe-type chondrodysplasia in a consanguineous Pakistani family. BMC Med Genet 2008;9:102. [Google Scholar] |
| 99. | Mumtaz S, Riaz HF, Touseef M, Basit S, Faiyaz Ul Haque M, Malik S. Recurrent mutation in CDMP1 in a family with Grebe chondrodysplasia: Broadening the phenotypic manifestation of syndrome in Pakistani population. Pak J Med Sci 2015;31:1542-4. [Google Scholar] |
| 100. | Khan S, Basit S, Khan MA, Muhammad N, Ahmad W. Genetics of human isolated acromesomelic dysplasia. Eur J Med Genet 2016;59:198-203. [Google Scholar] |
| 101. | Capellini TD, Chen H, Cao J, Doxey AC, Kiapour AM, Schoor M, et al. Ancient selection for derived alleles at a GDF5 enhancer influencing human growth and osteoarthritis risk. Nat Genet 2017;49:1202-10. [Google Scholar] |
| 102. | Pregizer SK, Kiapour AM, Young M, Chen H, Schoor M, Liu Z, et al. Impact of broad regulatory regions on Gdf5 expression and function in knee development and susceptibility to osteoarthritis. Ann Rheum Dis 2018;77:450. [Google Scholar] |
| 103. | Harada M, Takahara M, Zhe P, Otsuji M, Iuchi Y, Takagi M, et al. Developmental failure of the intra-articular ligaments in mice with absence of growth differentiation factor 5. Osteoarthritis Cartilage 2007;15:468-74. [Google Scholar] |
| 104. | Faiyaz-Ul-Haque M, Ahmad W, Zaidi SH, Haque S, Teebi AS, Ahmad M, et al. Mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene in a kindred affected with fibular hypoplasia and complex brachydactyly (DuPan syndrome). Clin Genet 2002;61:454-8. [Google Scholar] |
| 105. | Langer LO Jr., Cervenka J, Camargo M. A severe autosomal recessive acromesomelic dysplasia, the Hunter-Thompson type, and comparison with the grebe type. Hum Genet 1989;81:323-8. [Google Scholar] |
| 106. | Storm EE, Huynh TV, Copeland NG, Jenkins NA, Kingsley DM, Lee SJ. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 1994;368:639-43. [Google Scholar] |
| 107. | Coleman CM, Tuan RS. Functional role of growth/differentiation factor 5 in chondrogenesis of limb mesenchymal cells. Mech Dev 2003;120:823-36. [Google Scholar] |
| 108. | Shwartz Y, Viukov S, Krief S, Zelzer E. Joint development involves a continuous influx of gdf5-positive cells. Cell Rep 2016;15:2577-87. [Google Scholar] |
| 109. | Loughlin J. Knee osteoarthritis, lumbar-disc degeneration and developmental dysplasia of the hip – An emerging genetic overlap. Arthritis Res Ther 2011;13:108. [Google Scholar] |
| 110. | Miyamoto Y, Mabuchi A, Shi D, Kubo T, Takatori Y, Saito S, et al. Afunctional polymorphism in the 5' UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat Genet 2007;39:529-33. [Google Scholar] |
| 111. | Southam L, Rodriguez-Lopez J, Wilkins JM, Pombo-Suarez M, Snelling S, Gomez-Reino JJ, et al. An SNP in the 5'-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum Mol Genet 2007;16:2226-32. [Google Scholar] |
| 112. | Zhang R, Yao J, Xu P, Ji B, Luck JV, Chin B, et al. Acomprehensive meta-analysis of association between genetic variants of GDF5 and osteoarthritis of the knee, hip and hand. Inflamm Res 2015;64:405-14. [Google Scholar] |
| 113. | Rouault K, Scotet V, Autret S, Gaucher F, Dubrana F, Tanguy D, et al. Evidence of association between GDF5 polymorphisms and congenital dislocation of the hip in a Caucasian population. Osteoarthritis Cartilage 2010;18:1144-9. [Google Scholar] |
| 114. | Hatzikotoulas K, Roposch A; DDH Case Control Consortium, Shah KM, Clark MJ, Bratherton S, et al. Genome-wide association study of developmental dysplasia of the hip identifies an association with GDF5. Commun Biol 2018;1:56. [Google Scholar] |
| 115. | Sadat-Ali M, Al-Habdan IM, Bubshait DA. Genetic influence in developmental dysplasia of the hip in Saudi Arabian children due to GDF5 polymorphism. Biochem Genet 2018;56:618-26. [Google Scholar] |
| 116. | Christians JK, de Zwaan DR, Fung SH. Pregnancy associated plasma protein A2 (PAPP-A2) affects bone size and shape and contributes to natural variation in postnatal growth in mice. PLoS One 2013;8:e56260. [Google Scholar] |
| 117. | Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med 2016;8:363-74. [Google Scholar] |
| 118. | Banaszak-Ziemska M, Niedziela M. PAPP-A2 a new key regulator of growth. Endokrynol Pol 2017;68:682-91. [Google Scholar] |
| 119. | Conover CA, Boldt HB, Bale LK, Clifton KB, Grell JA, Mader JR, et al. Pregnancy-associated plasma protein-A2 (PAPP-A2): Tissue expression and biological consequences of gene knockout in mice. Endocrinology 2011;152:2837-44. [Google Scholar] |
| 120. | Shi D, Sun W, Xu X, Hao Z, Dai J, Xu Z, et al. Areplication study for the association of rs726252 in PAPPA2 with developmental dysplasia of the hip in Chinese Han population. Biomed Res Int 2014;2014:979520. [Google Scholar] |
| 121. | Favier B, Dollé P. Developmental functions of mammalian Hox genes. Mol Hum Reprod 1997;3:115-31. [Google Scholar] |
| 122. | Cobb J, Duboule D. Comparative analysis of genes downstream of the hoxd cluster in developing digits and external genitalia. Development 2005;132:3055-67. [Google Scholar] |
| 123. | Alvarado DM, McCall K, Hecht JT, Dobbs MB, Gurnett CA. Deletions of 5' HOXC genes are associated with lower extremity malformations, including clubfoot and vertical talus. J Med Genet 2016;53:250-5. [Google Scholar] |
| 124. | Zakany J, Duboule D. The role of Hox genes during vertebrate limb development. Curr Opin Genet Dev 2007;17:359-66. [Google Scholar] |
| 125. | Nelson CE, Morgan BA, Burke AC, Laufer E, DiMambro E, Murtaugh LC, et al. Analysis of Hox gene expression in the chick limb bud. Development 1996;122:1449-66. [Google Scholar] |
| 126. | Tickle C. Patterning systems – From one end of the limb to the other. Dev Cell 2003;4:449-58. [Google Scholar] |
| 127. | Jiang J, Ma HW, Lu Y, Wang YP, Wang Y, Li QW, et al. Transmission disequilibrium test for congenital dislocation of the hip and HOXB9 gene or COL1AI gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2003;20:193-5. [Google Scholar] |
| 128. | Rouault K, Scotet V, Autret S, Gaucher F, Dubrana F, Tanguy D, et al. Do HOXB9 and COL1A1 genes play a role in congenital dislocation of the hip? Study in a Caucasian population. Osteoarthritis Cartilage 2009;17:1099-105. [Google Scholar] |
| 129. | Schulte-Merker S, van Eeden FJ, Halpern ME, Kimmel CB, Nüsslein-Volhard C. No tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development 1994;120:1009-15. [Google Scholar] |
| 130. | Isaac A, Rodriguez-Esteban C, Ryan A, Altabef M, Tsukui T, Patel K, et al. Tbx genes and limb identity in chick embryo development. Development 1998;125:1867-75. [Google Scholar] |
| 131. | Naiche LA, Papaioannou VE. Loss of tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development 2003;130:2681-93. [Google Scholar] |
| 132. | Alvarado DM, Aferol H, McCall K, Huang JB, Techy M, Buchan J, et al. Familial isolated clubfoot is associated with recurrent chromosome 17q23.1q23.2 microduplications containing TBX4. Am J Hum Genet 2010;87:154-60. [Google Scholar] |
| 133. | Bongers EM, Duijf PH, van Beersum SE, Schoots J, Van Kampen A, Burckhardt A, et al. Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet 2004;74:1239-48. [Google Scholar] |
| 134. | Naiche LA, Papaioannou VE. Tbx4 is not required for hindlimb identity or post-bud hindlimb outgrowth. Development 2007;134:93-103. [Google Scholar] |
| 135. | Wang K, Shi D, Zhu P, Dai J, Zhu L, Zhu H, et al. Association of a single nucleotide polymorphism in Tbx4 with developmental dysplasia of the hip: A case-control study. Osteoarthritis Cartilage 2010;18:1592-5. [Google Scholar] |
| 136. | Hu Y, Vinayagam A, Nand A, Comjean A, Chung V, Hao T, et al. Molecular interaction search tool (MIST): An integrated resource for mining gene and protein interaction data. Nucleic Acids Res 2018;46:D567-D574. [Google Scholar] |
| 137. | Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The geneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 2010;38:W214-20. [Google Scholar] |
| 138. | Franz M, Rodriguez H, Lopes C, Zuberi K, Montojo J, Bader GD, et al. GeneMANIA update 2018. Nucleic Acids Res 2018;46:W60-4. [Google Scholar] |
Fulltext Views
4,135
PDF downloads
1,160




