
Translate this page into:
Orthopedic manifestations of congenital muscular dystrophy subtypes in children: Emerging signatures need consolidation: a scoping review

*Corresponding author: Tamer A. El-Sobky, Department of Orthopaedic Surgery, Division of Paediatric Orthopaedics, Faculty of Medicine, Ain Shams University, Cairo, Egypt. tamer.ahmed@med.asu.edu.eg
-
Received: ,
Accepted: ,
Supplementary material available online at https://doi.org/10.25259/JMSR_229_2023
Abstract
Our objective was to screen the literature on congenital muscular dystrophy (CMD) children/adolescents regarding the extent/nature of reporting orthopedic manifestations/deformities and to assess its appropriateness in informing clinical practice/research. We searched PubMed for original research on orthopedic surgical/non-surgical manifestations of CMD. Eligible articles needed to focus on orthopedic manifestations/deformities as one of the study objectives with no restrictions on study designs/types or search period. Eight hundred and thirty articles were initially identified and screened. Following the exclusion of 501 articles for disagreeing titles/abstracts, 329 were available for eligibility assessment. Two hundred and fifty-five articles were excluded for reasons. Of one hundred articles, 15 were captured manually and 11 through pre-submission searches, with 1078 patients included in the final analysis. The most common subtype was Laminin alpha-2 (LAMA2)-relatedCMD. Orthopedic manifestations of CMD are generally underreported and insufficiently detailed. There is reliable evidence that accurate reporting of orthopedic manifestations can be a valuable clinical supplement to the complex differential diagnosis process in collagen VI-related CMD, LAMA2-related-CMD, LMNA-related-CMD, and SEPN1-related CMD (SELENON). For alpha dystroglycan-related CMD, there is insufficient information to delineate a subtype-specific pattern. There is emerging evidence that reporting spine surgery outcomes may facilitate orthopedic decision making. The greatest clinical/research utility was provided by articles with longitudinal, comprehensive, and correlative reporting of larger cohorts. Detailed reporting of the orthopedic phenotype of CMD in future research may further uncover its diagnostic potential.
Keywords
Collagen type VI-related dystrophies
Laminopathies
LAMA2-related congenital muscular dystrophy
Rigid spine muscular dystrophy 1
SELENON-related myopathies
Walker-Warburg syndrome

INTRODUCTION
Congenital muscular dystrophy (CMD) spans a broad category of childhood-onset genetic muscle diseases that typically manifest in varying degrees of progressive muscle weakness, floppiness, and delayed motor milestones with no definite cure. The reported prevalence rates of CMD population studies range from 0.017/100,000 to 2.5/100,000.[1-3] However, CMD subtypes express remarkable diversity regarding clinical presentation, causative genetic mutations, muscle immunostaining, brain MRI findings,[4-6] muscle MRI findings,[7] especially whole-body muscle MRI,[8,9] associated cardiomyopathy,[10] and abnormalities in serum creatine kinase levels.[11] The classification of CMD is evolving due to the expanding and complex genotype-phenotype correlations. Congenital muscular dystrophies can be classified according to the type of structural muscle protein deficiency, causative gene mutation, and resultant phenotype/clinical profile. Broadly, the CMD classification includes (A) Laminin alpha-2 (LAMA2) or early-onset LAMA2- related CMD. Since LAMA2 isoform consists of chains α2, β1, and γ1, it is also labeled (laminin-211). Pathogenic mutations in the LAMA2 gen e cause early-onset LAMA2-related CMD. It was formerly known as merosin-deficient CMD type 1A and (B) alpha dystroglycan (DG)-related dystrophies. Pathogenic mutations in an array of genes – individually or in combination – cause Walker–Warburg syndrome, Fukuyama CMD, and muscle-eye-brain disease, among others and (C) Collagen VI-related dystrophies/CMD. Pathogenic mutations in COL6A1, COL6A2, and COL6A3 genes cause severe Ullrich, intermediate, and Bethlem CMD subtypes.[11-13] Miscellaneous subtypes of CMD include rigid spine syndrome or SEPN1-related myopathy caused by pathogenic mutations in the SEPN1 gene (SELENON; 606210). Lamin A/C (LMNA) or LMNA-related CMD is caused by pathogenic mutations in the LMNA gene.[14-18] This subtype belongs to a broader group known as laminopathies, which includes Emery-Dreifuss muscular dystrophy[14,19] and limb-girdle muscular dystrophy type 1B.[14] Marked cardiopulmonary insufficiency can occur in CMD.[12,20]
The CMD children share clinical manifestations common to all CMD subtypes, namely, neonatal or infantile muscle weakness, hypotonia, decreased spontaneous movements, and developmental motor delay. Disease progression eventually results in generalized contractures of the extremities and spine and compromised quality of life.[12,13,20] Conversely, certain disease-specific features can help differentiate the CMD subtypes. For example, the universal presence of white matter changes on the T2 signal of brain MRI with or without associated neurological manifestations[4,6,21] and the recent introduction of a characteristic disease signature on whole-body muscle MRI[8,9] can serve as valuable clues to the diagnosis of early-onset LAMA2-related CMD. However, overlapping and atypical clinical presentations of CMD subtypes [12,13,20] and other childhood-onset genetic muscle diseases such as congenital myopathies[20,22] can cause diagnostic challenges. For example, L-CMD and Emery-Dreifuss muscular dystrophy[15-18] should be strongly considered in the differential diagnosis of early-onset LAMA2-related CMD. In this respect, their presenting motor and orthopedic manifestations, disease severity, and prognosis bear a considerable resemblance to early-onset LAMA2-related CMD.[23,24]
Unlike Duchenne muscular dystrophy,[25-27] the orthopedic manifestations and outcomes of CMD subtypes have neither been widely researched nor precisely characterized in the literature.[23,28,29] Further, accurate characterization of orthopedic manifestations in CMD subtypes and their differential diagnosis may help resolve unexplained genotypephenotype correlations respecting overlapping clinical presentations, novel genes or multiple gene associations with disease, single gene association with multiple diseases, and clinically typical cases with negative genetic testing.[14,15,30,31] We assumed that delineating the pattern of orthopedic involvement in various CMD subtypes can improve diagnostic accuracy and provide treating physiatrists, physical therapists, and orthopedic surgeons with valuable clues as to what muscle groups one should prioritize in the treatment plan and what treatment endpoints should one aim at and so forth. This topic has not been addressed previously in the literature. This scoping review aimed to screen the current literature on CMD children and adolescents, respecting the extent and precision of reporting orthopedic manifestations and its appropriateness in informing clinical practice and research.
MATERIAL AND METHODS
Identifying research questions
This scoping review was carried out in accordance with the guidelines of the PRISMA extension for scoping reviews.[32,33]
The goal of this scoping review was broken down into three research subquestions, namely;
How often are orthopedic manifestations reported in publications on CMD children and adolescents?
How precise is the reporting of the orthopedic manifestations, for example, completeness and accuracy?
How appropriate is it in informing clinical practice and research?
The above specifics of reporting orthopedic manifestations were weighed against the standard components of orthopedic examination whenever applicable. That is history-taking and orthopedic couch examination with special emphasis on deformity/contracture characteristics. Gait analysis and motor development were not directly targeted. For clarity, the terms orthopedic manifestations and orthopedic deformities/contractures will often be used synonymously across the manuscript.
Search strategy
We searched the publications in MEDLINE through PubMed advanced search builder https://pubmed.ncbi.nlm.nih.gov/advanced/#. We performed the initial search in November 2021, and an additional search was performed on April 23, 2022 before manuscript submission to ensure the identified literature was up to date. The age filter was set to child: Birth-18 years and species set to humans. We did not include additional search filters. Our search strategy comprised phrases, index words, and Medical Subject Headings pertinent to pathology, that is, CMD subtypes. We used the following words to indicate the three main CMD subtypes: CMD, Walker-Warburg syndrome, LAMA2, merosin, Bethlem, and Ullrich. We used other words in separate searches to indicate the miscellaneous and less common CMD subtypes, namely, rigid spine syndrome, SEPN1-related myopathy, LMNA-related CMD, and laminopathies. The search objective was mainly focused on recall. We screened the titles and abstracts of the initially identified articles. After making necessary omissions, we assessed the remaining articles for potential inclusion.
Moreover, we obtained the full text of the final included articles [Supplementary File S1]. A flowchart of the literature extraction process is shown [Figure 1]. We checked the reference lists of the relevant articles and reviews for additional eligible articles. We manually checked similar articles and cited by functions regarding these relevant articles.

- Flowchart of the literature extraction process. An updated search was performed before manuscript submission to ensure the identified literature was current. Detailed description of excluded articles with reasons can be found in Supplementary File S1.
Eligibility criteria
Articles were included if they represented original/primary research and addressed orthopedic manifestations in any subtype of CMD in a descriptive or intervention (surgical or non-surgical) setting. To be included, the article needed to focus clearly on reporting the clinical orthopedic examination/manifestations as one of the study objectives. This can occur solely or among other clinical manifestations. Simple/broad or binary referral to the presence or absence of orthopedic manifestations was not included in the analysis. This scoping review was primarily exploratory and descriptive regarding the evidence for reporting orthopedic manifestations. Thus, we considered all study designs, for example, (longitudinal vs. cross-sectional/prospective vs. retrospective) and all research types (case reports vs. case series) eligible for inclusion. Articles were also included if they investigated CMD exclusively or as a constituent of a larger cohort of genetic muscle diseases. We excluded non-peer-reviewed material, experimental or preclinical articles, and those reporting exclusively on genetic muscle diseases other than CMD as congenital myopathies.
Data extraction and synthesis of results
We extracted elementary data elements, namely, study designs and research types, patient populations (CMD subtype), and authors’ disciplines of included articles. In this regard, we were particularly interested in identifying authors or coauthors in the discipline of musculoskeletal/orthopedics, namely, rehabilitation physicians, physiotherapists, orthotists, and orthopedic surgeons. In addition, we extracted data pertaining to the research questions, namely, (1) frequency and nature of reporting, which included qualitative or categorical reporting or quantitative as goniometric joint measurements, etc., (2) precision, which included completeness and accuracy of reporting of orthopedic manifestations, and (3) appropriateness in informing clinical practice and research. The authors performed a pilot test on screening the titles and abstracts of the initially identified articles and another on data extraction/charting of the included full-text articles. Disagreements between authors were settled by joint meetings. The final review and charting of the above data items were performed by author TAE, whereas the rest of the authors verified the data extraction/charting. We did not critically appraise the sources of evidence – articles – included in this scoping review.
RESULTS
Characteristics of sources of evidence
The 100 final articles included peer-reviewed descriptive observational research, namely, case reports of one or two patients, 47 (47%) articles or case series of affected families or unrelated patients, and 53 (53%) articles. The diagnosis was verified with the aid of genetic analysis and/or muscle biopsy immunostaining supplemented by clinical presentation and brain MRI or neuromuscular electrophysiological investigations.
Results of individual sources of evidence
The initial search for the three main CMD subtypes and the two additional searches for LMNA -related CMD, a subtype of laminopathies and SEPN1 -related myopathies/CMD or rigid spine syndrome identified 830 articles, and the titles and abstracts of which were screened for any irrelevant articles. Following the exclusion of 501 irrelevant articles, 329 remaining ones were assessed for eligibility criteria. Additional articles were excluded with reasons (n = 255) either due to the clinical content, and consequently, the orthopedic was absent or near absent as a study objective, for example, focused on genetic analysis, muscle biopsy/immunostaining, or muscle/brain MRI features and so forth or because the articles were primarily focused on a specific non-orthopedic clinical parameter, for example, cardiac, anesthetic complications or because the article’s clinical orthopedic content was not reported as per eligibility criteria, either solely or as one of the constituents of clinical manifestations, among other reasons [Supplementary File S1 and S2]. These latter exclusions necessitated retrieval and investigation of the full-text articles, six of which were irretrievable. Among the latter exclusions were five articles that reported negative/absent orthopedic manifestations. Although this was deemed adequate reporting, they did not technically allow for evaluation of the precision of reporting. Fifteen more articles were added through manual search, as they fit the inclusion criteria. Thus, the final number of included articles in the current review was 100.[14,23,28,34-130] Of the 100 articles, two[95,96] were captured through an additional search performed on April 23, 2022, before manuscript submission to ensure the identified literature was up to date [Supplementary File S1]. Another nine articles[122-130] were captured through another pre-submission-focused search performed on May 19, 2022 [Supplementary File S1]. The latter complementary search was intended specifically to enhance the recall of articles on Walker-Warburg syndrome (alpha DG -related CMD). The total number of patients enrolled in these articles was 1078, distributed as follows: LAMA2-related CMD 399 (37%) patients, collagen VI-related CMD 132 (12%), alpha DG -related CMD 75 (7%), LMNA-related rigid spine CMD/syndrome 196 (18%), and LMNA -related CMD 276 (26%). A flowchart of the literature extraction process is shown in Figure 1. Of the final included articles, 94 (94%) addressed clinical – non-surgical – orthopedic aspects of CMD, and 6 (6%) addressed orthopedic surgical interventions.[35-37,73,84,110] Only 26 (26%) of articles included authors affiliated to musculoskeletal disciplines, medicine or surgery; orthopedic surgery 18 articles,[28,35-37,39,40,42,48,53,73,84,87,104,105,110,117,119,125] rehabilitation medicine/therapy 7,[44,83,91,94,97,103,112] and kinetic therapy 1.[43]
Synthesis of results
Data items and extraction (charting) of included articles are shown in [Supplementary Tables S1-3] per CMD subtype. Overall, there seems to be a fairly consistent pattern of orthopedic manifestations for most of the CMD subtypes [Figure 2]. Most of the 329 articles retrieved for eligibility assessment were excluded 255 (77.5%) despite being pertinent to clinical research on CMD. The full text of the final included articles was searched for precision in reporting orthopedic manifestations (deformities or contractures) in CMD regarding anatomic localization of deformities, severity grading, symmetry, range of motion features, age at onset and course of deformity, and additional relevant information. The majority of included case series articles did not report orthopedic contractures in a correlative manner, namely, correlations between orthopedic contractures on the one side and patient’s genotype, distribution and onset of muscle weakness/wasting, general motor function, and so forth on the other side. There were notable exceptions.[14,87,94] Although many of the included articles reported the overall disease progression and anatomic distribution of muscle weakness, few reported the progression and distribution of orthopedic contractures particularly. There were notable exceptions.[23,94] The overall orthopedic indicators of CMD subtypes are shown in Figure 2. A proposed flowchart for comprehensive eporting of orthopedic deformities in CMD children is presented in Figure 3. We successfully contacted the authors of the included articles[111] to retrieve relevant supplementary files, which were inaccessible in the PDF versions. We unsuccessfully contacted the authors of an eligible yet excluded article[131] to retrieve supplementary files, which contained the clinical spectrum of alpha DG -related CMD in a large cohort of Chinese patients.
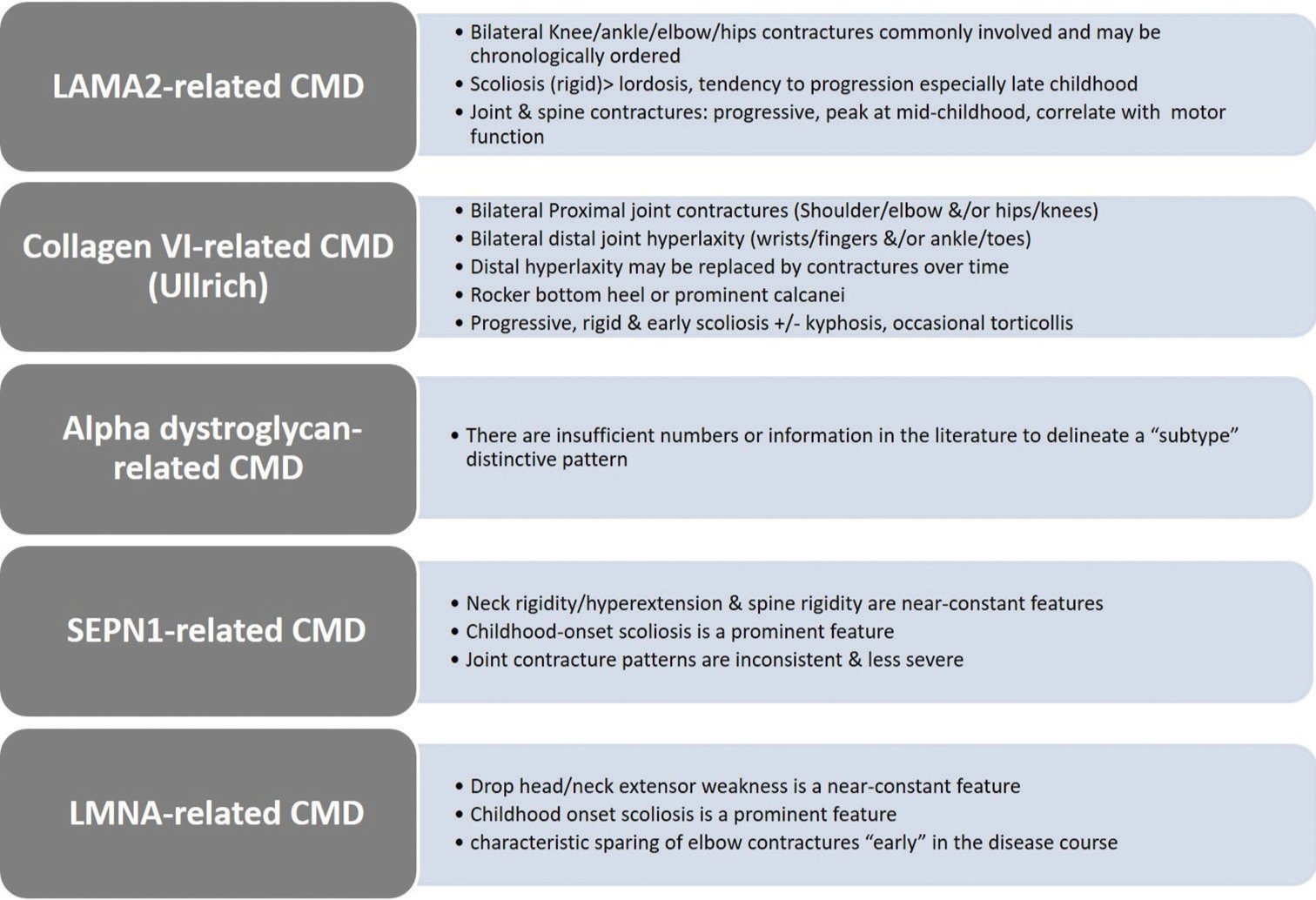
- Graphical abstract image: General orthopedic indicators of congenital muscular dystrophy (CMD) subtypes. Indicators are based on the best available evidence and are not necessarily all-inclusive or pathognomonic. Collagen VI-related CMD indicators are most applicable to the Ullrich subtype.
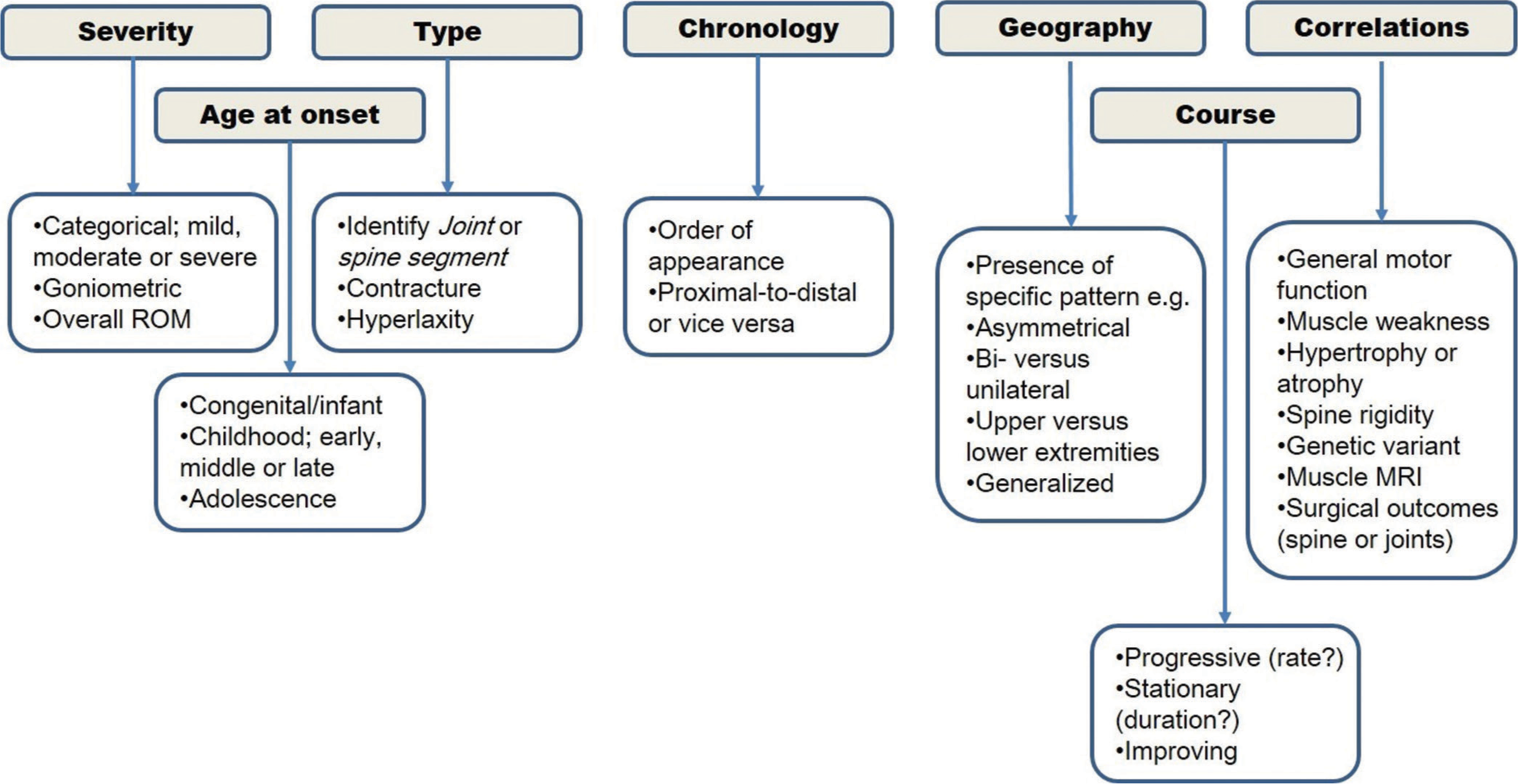
- Proposed flowchart for comprehensive reporting of orthopedic deformities in congenital muscular dystrophy children.
DISCUSSION
The fact that the vast majority of articles retrieved for eligibility assessment were excluded despite being pertinent to clinical research on CMD denotes that orthopedic manifestations in CMD are underreported both quantitatively and qualitatively. That is, these articles did not contain clinically meaningful or important orthopedic content to warrant investigation. The final articles included in our study (21.5%) yielded various qualities of reporting orthopedic manifestations. For example, articles on collagen VI CMD, Ullrich subtype reported a fairly consistent and characteristic pattern of orthopedic manifestations. A notable example is the characteristic coexistence of large/proximal joint contractures and small/distal joint laxity in these patients.[38-40,42,45] A similarly characteristic pattern of orthopedic manifestations was reported in a large cohort of LAMA2-related CMD, which established in addition genotype/orthopedic-type correlations[87] among others.[28,94] Additional emerging characteristic orthopedic patterns were demonstrated in large cohorts of SEPN1-related subtypes of CMD or rigid spine syndrome[97] and LMNA-related CMD.[112,113] Such an orthopedic signature could serve as a valuable and handy clinical differential diagnostic aid, especially if established in other subtypes of CMD. Orthopedic manifestations, particularly the chronological order of appearance of joint contractures, symmetry/laterality and degree of rigidity, need accurate reporting. For example, early appearance of elbow contractures was found to be a potentially useful diagnostic sign in patients with Emery-Dreifuss muscular dystrophy, in contrast to LMNA-related CMD and limb-girdle muscular dystrophy type 1B where they appeared late in the disease course[114,115,117] and so forth. The favorable intermediate-term surgery outcomes for spinal deformities secondary to CMD may prove to be another valuable decision-making tool and an indicator of responsiveness to orthopedic surgery.[35-37,73,84]
Orthopedic manifestations of neuromuscular diseases
The motor and orthopedic manifestations of genetic neuromuscular diseases are valuable clinical tools allowing for refinement of differential diagnosis,[132,133] identification of age- and disease-specific orthopedic and rehabilitation treatment endpoints,[29,134-137] informed surgical decision-making,[138] and assessment of efficacy of experimental therapeutics and improvement of clinical trial designs.[139] For example, emerging evidence suggests that familiarity with orthopedic manifestations can help in the differential diagnosis of Charcot-Marie-Tooth subtypes and related genetic polyneuropathies[132] and fascioscapulohumeral muscular dystrophy[133] and guide their orthopedic and rehabilitation management plans. Additional studies underscored the importance of orthopedic manifestations to the surgical decision-making in myotonic dystrophy[140] and to the monitoring of enzyme replacement therapy in early-onset Pompe disease.[137,141] The accurate description of the orthopedic manifestations of CMD subtypes[28,38-40] is particularly important to low-resource countries where diagnosis and subsequent referral to specialized centers depend highly on the physician’s clinical judgment.[142]
Although experimental research on CMD subtypes is promising,[143-147] early diagnosis can facilitate supportive and system-specific management per recommended disease standards of care.[12,148] Relatedly, neglected spine deformities can accentuate cardiac[21,149-151] and pulmonary[20,152] complications.
Strengths and limitations
The strength of our study is that it mapped an underreported yet central feature in CMD children, namely, orthopedic manifestations.[148,149,153-156] Accurate and systematic reporting can provide valuable insights into the mechanisms of disease causation of the wide mutation/variant spectrum CMD subtypes. It can also establish correlative relationships between gene variants and the diverse phenotype presentations of CMD subtypes and similar genetic muscle diseases with result improvement in the genetic/molecular diagnostic yield.[22,30,31,157] The elucidation of the orthopedic manifestations in future reports can expand the phenotype/clinical profile of CMD subtypes, which can help clinicians diagnose CMD subtypes with more precision. Our study has limitations. We searched only one database. Given the ultra-specialized nature of CMD, we assumed that most research on it would be published in reputable PubMed-indexed journals. Consequently, we believe that our search should have captured at least the main bulk of relevant articles.
Further, the recall and precision of a search strategy depend on multiple variables such as the operational specifics and degree of indexing inaccuracies or delays of the electronic database and the contribution of a health information librarian to the search design among others. However, we believe that the ample amount of literature recall regarding the included number of articles and patients in this analysis has provided fairly representative material to justify meaningful conclusions, at least as far as a scoping review is concerned.
CONCLUSION
The clinical and surgical orthopedic manifestations of CMD subtypes in children and adolescents are generally underreported and insufficiently detailed in the literature. However, there is reliable evidence that accurate reporting of orthopedic manifestations can be a valuable clinical supplement to the complex differential diagnosis process of children and adolescents with collagen VI-related, LAMA2-related, LMNA-related, and SEPN1related CMD (SELENON). For alpha DG -related CMD, there is insufficient information to delineate a subtype-specific pattern. There is emerging evidence that reporting spine surgery outcomes may facilitate orthopedic decision-making in CMD. Predictors of high clinical and research utility were articles with longitudinal, comprehensive, and correlative reporting of larger cohorts. Detailed reporting of the orthopedic phenotype of CMD in future research may further uncover its diagnostic potential.
What does this paper add?
Orthopedic manifestations of congenital muscular dystrophy (CMD) in children/adolescents are generally underreported and insufficiently detailed.
There is reliable evidence that accurate reporting of orthopedic manifestations can be a valuable clinical supplement to differential diagnosis in patients with collagen VI-related, LAMA2-related, LMNA-related, and SEPN1-related CMD (SELENON).
For alpha dystroglycan-related CMD, there is insufficient information to delineate a subtype-specific pattern.
There is emerging evidence that reporting spine surgery outcomes may assist in orthopedic decision-making.
The greatest clinical/research utility was provided by articles with longitudinal, comprehensive, and correlative reporting of larger cohorts.
Comprehensive reporting of orthopedic manifestations is recommended in future research.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article and its supplementary information files.
AUTHORS’ CONTRIBUTIONS
TAE conceptualized and designed the study. TAE, HA, SM, and JA contributed to data acquisition, analysis and interpretation. TAE drafted the manuscript. All authors revised the manuscript critically for important intellectual content and approved the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
ETHICAL APPROVAL
The authors confirm that this review had been prepared in accordance with COPE roles and regulations. Given the nature of the review, the IRB review was not required.
DECLARATION OF PATIENT CONSENT
Patient’s consent was not required as there are no patients in this study.
USE OF ARTIFICIAL INTELLIGENCE (AI) ASSISTED TECHNOLOGY FOR MANUSCRIPT PREPARATION
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
CONFLICTS OF INTEREST
There are no conflicting relationships or activities.
FINANCIAL SUPPORT AND SPONSORSHIP
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- Congenital muscular dystrophies in China. Clin Genet. 2019;96:207-15.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence of congenital muscular dystrophy in Italy: A population study. Neurology. 2015;84:904-11.
- [CrossRef] [PubMed] [Google Scholar]
- Neuromuscular disorders in childhood: A descriptive epidemiological study from western Sweden. Neuromuscul Disord. 2000;10:1-9.
- [CrossRef] [PubMed] [Google Scholar]
- Epilepsy in LAMA2-related muscular dystrophy: A systematic review of the literature. Seizure. 2021;91:425-36.
- [CrossRef] [PubMed] [Google Scholar]
- New MRI findings in Fukuyama congenital muscular dystrophy: Brain stem and venous system anomalies. AJNR Am J Neuroradiol. 2020;41:1094-8.
- [CrossRef] [PubMed] [Google Scholar]
- Epilepsy in LAMA2-related muscular dystrophy: An electro-clinico-radiological characterization. Epilepsia. 2020;61:971-83.
- [CrossRef] [PubMed] [Google Scholar]
- Characteristic muscle signatures assessed by quantitative MRI in patients with Bethlem myopathy. J Neurol. 2020;267:2432-42.
- [CrossRef] [PubMed] [Google Scholar]
- Whole-body muscle MRI characteristics of LAMA2-related congenital muscular dystrophy children: An emerging pattern. Neuromuscul Disord. 2021;31:814-23.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic interest of whole-body MRI in early-and late-onset LAMA2 muscular dystrophies: A large international cohort. J Neurol. 2022;269:2414-29.
- [CrossRef] [PubMed] [Google Scholar]
- Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-α2 chain-deficient congenital muscular dystrophy. Appl Clin Genet. 2019;12:113-30.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital muscular dystrophy: From muscle to brain. Ital J Pediatr. 2016;42:78.
- [CrossRef] [PubMed] [Google Scholar]
- Evidence-based guideline summary: Evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular and Electrodiagnostic Medicine. Neurology. 2015;84:1369-78.
- [CrossRef] [PubMed] [Google Scholar]
- Fukuyama congenital muscular dystrophy In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, eds. GeneReviews®. Seattle WA: University of Washington 1993-2023; 2006. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1206 [Last accessed on 2019 Jul 03]
- [Google Scholar]
- International retrospective natural history study of LMNA-related congenital muscular dystrophy. Brain Commun. 2021;3:fcab075.
- [Google Scholar]
- Lamin A/C assembly defects in LMNA-congenital muscular dystrophy is responsible for the increased severity of the disease compared with Emery-Dreifuss muscular dystrophy. Cells. 2020;9:844.
- [CrossRef] [PubMed] [Google Scholar]
- Dropped head related lamin A/C associated congenital muscular dystrophy case; previously defined as emerydreifuss muscular dystrophy. Turk J Pediatr. 2020;62:130-5.
- [CrossRef] [PubMed] [Google Scholar]
- A novel LMNA mutation identified in a Japanese patient with LMNA-associated congenital muscular dystrophy. Hum Genome Var. 2018;5:19.
- [CrossRef] [PubMed] [Google Scholar]
- Lamin-Related congenital muscular dystrophy alters mechanical signaling and skeletal muscle growth. Int J Mol Sci. 2020;22:306.
- [CrossRef] [PubMed] [Google Scholar]
- Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2020;61:436-48.
- [CrossRef] [PubMed] [Google Scholar]
- LAMA2 muscular dystrophy. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, eds. GeneReviews®. Seattle, WA: University of Washington 1993-2023; 2012. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK97333 [Last accessed on 2020 Sep 17]
- [Google Scholar]
- Atypical phenotype in two patients with LAMA2 mutations. Neuromuscul Disord. 2014;24:419-24.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic cause of heterogeneous inherited myopathies in a cohort of Greek patients. Mol Genet Metab Rep. 2020;25:100682.
- [CrossRef] [PubMed] [Google Scholar]
- LAMA2-related muscular dystrophy: Natural history of a large pediatric cohort. Ann Clin Transl Neurol. 2020;7:1870-82. Erratum Ann Clin Transl Neurol 2021;8,1571
- [CrossRef] [PubMed] [Google Scholar]
- LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum Mutat. 2018;39:1314-37.
- [CrossRef] [PubMed] [Google Scholar]
- Rehabilitation management of the patient with Duchenne muscular dystrophy. Pediatrics. 2018;142(Suppl 2):S17-33.
- [CrossRef] [PubMed] [Google Scholar]
- Orthopedic and surgical management of the patient with Duchenne muscular dystrophy. Pediatrics. 2018;142(Suppl 2):S82-9.
- [CrossRef] [PubMed] [Google Scholar]
- Ambulatory Duchenne muscular dystrophy children: Cross-sectional correlation between function, quantitative muscle ultrasound and MRI. Acta Myol. 2022;10:1-14.
- [CrossRef] [Google Scholar]
- Whole-body muscle magnetic resonance imaging characteristics of children with merosin-deficient congenital muscular dystrophy. Dev Med Child Neurol. 2018;60:94-5.
- [CrossRef] [Google Scholar]
- Management of motor rehabilitation in individuals with muscular dystrophies. 1st Consensus Conference report from UILDM-Italian Muscular Dystrophy Association (Rome, January 25-26 2019) Acta Myol. 2021;40:72-87.
- [Google Scholar]
- Clinical and genomic evaluation of 207 genetic myopathies in the Indian subcontinent. Front Neurol. 2020;11:559327.
- [CrossRef] [PubMed] [Google Scholar]
- The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet. 2017;62:243-52.
- [CrossRef] [PubMed] [Google Scholar]
- PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and explanation. Ann Intern Med. 2018;169:467-73.
- [CrossRef] [PubMed] [Google Scholar]
- Updated methodological guidance for the conduct of scoping reviews. JBI Evid Synth. 2020;18:2119-26.
- [CrossRef] [PubMed] [Google Scholar]
- Ullrich congenital muscular dystrophy and Bethlem myopathy: Clinical and genetic heterogeneity. Arq Neuropsiquiatr. 2005;63:785-90.
- [CrossRef] [PubMed] [Google Scholar]
- Spinal correction in patients with Fukuyama congenital muscular dystrophy. J Orthop Sci. 2017;22:658-64.
- [CrossRef] [PubMed] [Google Scholar]
- Surgical correction of spinal deformity in patients with congenital muscular dystrophy. J Orthop Sci. 2010;15:493-501.
- [CrossRef] [PubMed] [Google Scholar]
- Spinal fusion in a patient with Fukuyama congenital muscular dystrophy. Brain Dev. 2017;39:613-6.
- [CrossRef] [PubMed] [Google Scholar]
- Ullrich congenital muscular dystrophy: Report of nine cases from India. Neurol India. 2009;57:41-5.
- [CrossRef] [PubMed] [Google Scholar]
- Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann Neurol. 2005;58:400-10.
- [CrossRef] [PubMed] [Google Scholar]
- Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci U S A. 2001;98:7516-21.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and histopathological study of merosin-deficient and merosin-positive congenital muscular dystrophy. Pediatr Dev Pathol. 2000;3:168-76.
- [CrossRef] [PubMed] [Google Scholar]
- Collagen VI involvement in Ullrich syndrome: A clinical, genetic, and immunohistochemical study. Neurology. 2002;58:1354-9.
- [CrossRef] [PubMed] [Google Scholar]
- Merosin-deficient congenital muscular dystrophy type 1A. Rom J Morphol Embryol. 2008;49:229-33.
- [Google Scholar]
- Congenital muscular dystrophy with rigid spine syndrome: A clinical, pathological, radiological, and genetic study. Ann Neurol. 2000;47:152-61.
- [CrossRef] [PubMed] [Google Scholar]
- Novel collagen VI mutations identified in Chinese patients with Ullrich congenital muscular dystrophy. World J Pediatr. 2014;10:126-32.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital muscular dystrophy with laminin alpha 2 chain deficiency: Identification of a new intermediate phenotype and correlation of clinical findings to muscle immunohistochemistry. Eur J Pediatr. 1996;155:968-76.
- [CrossRef] [PubMed] [Google Scholar]
- Merosin-positive congenital muscular dystrophy in two siblings with cataract and slight mental retardation. Brain Dev. 1999;21:274-8.
- [CrossRef] [PubMed] [Google Scholar]
- Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: Phenotype analysis of 11 families linked to the COL6 loci. Neuropediatrics. 2004;35:103-12.
- [CrossRef] [PubMed] [Google Scholar]
- Compound heterozygous POMGNT1 mutations leading to muscular dystrophy-dystroglycanopathy type A3: A case report. BMC Pediatr. 2019;19:98.
- [CrossRef] [PubMed] [Google Scholar]
- Ullrich's congenital atonic sclerotic muscular dystrophy. A case report. J Neurol. 1989;236:108-10.
- [CrossRef] [PubMed] [Google Scholar]
- Large genomic deletions: A novel cause of Ullrich congenital muscular dystrophy. Ann Neurol. 2011;69:206-11.
- [CrossRef] [PubMed] [Google Scholar]
- A novel de novo COL6A1 mutation emphasizes the role of intron 14 donor splice site defects as a cause of moderate-progressive form of ColVI myopathy-a case report and review of the genotypephenotype correlation. Folia Neuropathol. 2017;55:214-20.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet. 2002;70:1446-58.
- [CrossRef] [PubMed] [Google Scholar]
- A novel variant in the COL6A1 gene causing Ullrich congenital muscular dystrophy in a consanguineous family: A case report. BMC Neurol. 2021;21:105.
- [CrossRef] [PubMed] [Google Scholar]
- Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology. 1995;45:2084-9.
- [CrossRef] [PubMed] [Google Scholar]
- New molecular mechanism for Ullrich congenital muscular dystrophy: A heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73:355-69.
- [CrossRef] [PubMed] [Google Scholar]
- Genetically confirmed patients with merosin-deficient congenital muscular dystrophy in China. Neuropediatrics. 2008;39:264-7.
- [CrossRef] [PubMed] [Google Scholar]
- Severe congenital muscular dystrophy in a LAMA2-mutated case. Pediatr Neurol. 2007;37:212-4.
- [CrossRef] [PubMed] [Google Scholar]
- Merosin-deficient congenital muscular dystrophy with mental retardation and cerebellar cysts, unlinked to the LAMA2, FCMD, MEB and CMD1B loci, in three Tunisian patients. Neuromuscul Disord. 2003;13:4-12.
- [CrossRef] [PubMed] [Google Scholar]
- Merosin-negative congenital muscular dystrophy: Report of five cases. J Pediatr Neurosci. 2015;10:346-9.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of a new locus for a peculiar form of congenital muscular dystrophy with early rigidity of the spine, on chromosome 1p35-36. Am J Hum Genet. 1998;62:1439-45.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the laminin alpha2-chain gene in two children with early-onset muscular dystrophy. Brain. 2000;123:31-41.
- [CrossRef] [PubMed] [Google Scholar]
- Substitution of a conserved cysteine-996 in a cysteine-rich motif of the laminin alpha2-chain in congenital muscular dystrophy with partial deficiency of the protein. Am J Hum Genet. 1996;58:1177-84.
- [Google Scholar]
- Congenital, hypotonic-sclerotic muscular dystrophy. J Med Genet. 1977;14:426-9.
- [CrossRef] [PubMed] [Google Scholar]
- A clinical and histological study of Ullrich's disease (congenital atonic-sclerotic muscular dystrophy) Neuropediatrics. 1981;12:197-208.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical features and molecular characterization of a patient with muscle-eye-brain disease: A novel mutation in the POMGNT1 gene. J Child Neurol. 2014;29:289-94.
- [CrossRef] [PubMed] [Google Scholar]
- Late onset scleroatonic familial myopathy (Ullrich disease): A study of two sibs. Am J Med Genet. 1988;31:933-42.
- [CrossRef] [PubMed] [Google Scholar]
- Laminin-alpha 2 chain (merosin M) is preserved in the Walker-Warburg syndrome. Neuropediatrics. 1996;27:279-80.
- [CrossRef] [PubMed] [Google Scholar]
- LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dystrophy. Neurology. 2004;63:1118-21.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha 2 (LAMA2) deficiency. Hum Mutat. 2003;21:103-11.
- [CrossRef] [PubMed] [Google Scholar]
- Merosin-deficient congenital muscular dystrophy in two siblings. Hong Kong Med J. 2004;10:423-6.
- [Google Scholar]
- Divergence of central nervous system involvement in 2 Italian sisters with congenital muscular dystrophy: A clinical and neuroradiological follow-up. Eur Neurol. 1995;35:230-5.
- [CrossRef] [PubMed] [Google Scholar]
- Cervical hyperextension treated by posterior spinal correction and fusion in a patient with Ullrich congenital muscular dystrophy: A case report. JBJS Case Connect. 2020;10:e0392.
- [CrossRef] [PubMed] [Google Scholar]
- A variant of congenital muscular dystrophy. Brain Dev. 2002;24:24-9.
- [CrossRef] [PubMed] [Google Scholar]
- A case of Ullrich's disease (Kongenitale, Atonisch-Sklerotische Muskeldystrophie) Brain Dev. 1979;1:61-7.
- [CrossRef] [PubMed] [Google Scholar]
- Mild clinical phenotype in a 12-year-old boy with partial merosin deficiency and central and peripheral nervous system abnormalities. Neuromuscul Disord. 1996;6:377-81.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital muscular dystrophy phenotype with neuromuscular spindles excess in a 5-year-old girl caused by HRAS mutation. Neuromuscul Disord. 2014;24:993-8.
- [CrossRef] [PubMed] [Google Scholar]
- Preserved merosin M-chain (or laminin-alpha 2) expression in skeletal muscle distinguishes Walker-Warburg syndrome from Fukuyama muscular dystrophy and merosin-deficient congenital muscular dystrophy. Neuropediatrics. 1995;26:148-55.
- [CrossRef] [PubMed] [Google Scholar]
- A Danish family with limb-girdle muscular dystrophy with autosomal dominant inheritance. Neuromuscul Disord. 1994;4:139-42.
- [CrossRef] [PubMed] [Google Scholar]
- Fukuyama-type congenital muscular dystrophy: A case report in the Japanese population living in Brazil. Acta Neurol Scand. 2002;106:117-21.
- [CrossRef] [PubMed] [Google Scholar]
- LAMA2 mRNA processing alterations generate a complete deficiency of laminin-alpha2 protein and a severe congenital muscular dystrophy. Neuromuscul Disord. 2008;18:137-45.
- [CrossRef] [PubMed] [Google Scholar]
- A homozygous COL6A2 intron mutation causes in-frame triple-helical deletion and nonsense-mediated mRNA decay in a patient with Ullrich congenital muscular dystrophy. Hum Genet. 2005;117:460-6.
- [CrossRef] [PubMed] [Google Scholar]
- Ullrich disease: Collagen VI deficiency: EM suggests a new basis for muscular weakness. Neurology. 2002;59:920-3.
- [CrossRef] [PubMed] [Google Scholar]
- Tailor-made management of thoracic scoliosis with cervical hyperextension in muscular dystrophy. Eur Spine J. 2018;27:264-9.
- [CrossRef] [PubMed] [Google Scholar]
- Merosin-positive congenital muscular dystrophy with mental retardation, microcephaly and central nervous system abnormalities unlinked to the Fukuyama muscular dystrophy and muscular-eye-brain loci: Report of three siblings. Neuromuscul Disord. 2001;11:570-8.
- [CrossRef] [PubMed] [Google Scholar]
- Fukutin gene retrotransposal insertion in a non-Japanese Fukuyama congenital muscular dystrophy (FCMD) patient. Am J Med Genet A. 2009;149A:2403-8.
- [CrossRef] [PubMed] [Google Scholar]
- Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort. Orphanet J Rare Dis. 2021;16:319.
- [CrossRef] [PubMed] [Google Scholar]
- Muscle magnetic resonance imaging in patients with various clinical subtypes of LMNA-related muscular dystrophy. Chin Med J (Engl). 2018;131:1472-9.
- [CrossRef] [PubMed] [Google Scholar]
- Variable disease severity in Saudi Arabian and Sudanese families with c.3924 + 2 T > C mutation of LAMA2. BMC Res Notes. 2011;4:534.
- [CrossRef] [PubMed] [Google Scholar]
- Exome sequencing detects compound heterozygous nonsense LAMA2 mutations in two siblings with atypical phenotype and nearly normal brain MRI. Neuromuscul Disord. 2019;29:376-80.
- [CrossRef] [PubMed] [Google Scholar]
- LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. 2008;74:502-12.
- [CrossRef] [PubMed] [Google Scholar]
- Laminin α2 deficiency-related muscular dystrophy mimicking Emery-Dreifuss and collagen vi related diseases. J Neuromuscul Dis. 2015;2:229-40.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and neuroimaging findings in two brothers with limb girdle muscular dystrophy due to LAMA2 mutations. Neuromuscul Disord. 2017;27:170-4.
- [CrossRef] [PubMed] [Google Scholar]
- Longitudinal changes in clinical outcome measures in COL6-related dystrophies and LAMA2-related dystrophies. Neurology. 2019;93:e1932-43.
- [CrossRef] [Google Scholar]
- Intrafamilial phenotypic variability of collagen vi-related myopathy due to a new mutation in the COL6A1 Gene. J Neuromuscul Dis. 2021;8:273-85.
- [CrossRef] [PubMed] [Google Scholar]
- A Revisited diagnosis of collagen VI related muscular dystrophy in a patient with a novel COL6A2 variant and 21q22.3 deletion. Neuropediatrics. 2020;51:445-9.
- [CrossRef] [PubMed] [Google Scholar]
- The clinical, histologic, and genotypic spectrum of SEPN1-related myopathy: A case series. Neurology. 2020;95:e1512-27.
- [CrossRef] [PubMed] [Google Scholar]
- SEPN1-related rigid spine muscular dystrophy. Indian J Pediatr. 2018;85:1033-4.
- [CrossRef] [PubMed] [Google Scholar]
- SEPN1-related myopathy in three patients: Novel mutations and diagnostic clues. Eur J Pediatr. 2016;175:1113-8.
- [CrossRef] [PubMed] [Google Scholar]
- Rigid spinal muscular dystrophy and rigid spine syndrome: Report of 7 children. J Child Neurol. 2014;29:1436-40.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and imaging findings in six cases of congenital muscular dystrophy with rigid spine syndrome linked to chromosome 1p (RSMD1) Neuromuscul Disord. 2002;12:631-8.
- [CrossRef] [PubMed] [Google Scholar]
- SEPN1-related myopathy: The importance of diagnosis and challenges to management of CMD in resource poor settings. Ann Indian Acad Neurol. 2021;24:955-6.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital muscular dystrophy and rigid spine syndrome. Neuropediatrics. 1983;14:97-101.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic heterogeneity of congenital muscular dystrophy with rigid spine syndrome. Neuromuscul Disord. 1999;9:376-82.
- [CrossRef] [PubMed] [Google Scholar]
- SEPN1-related myopathies: Clinical course in a large cohort of patients. Neurology. 2011;76:2073-8.
- [CrossRef] [PubMed] [Google Scholar]
- A single homozygous point mutation in a 3'untranslated region motif of selenoprotein N mRNA causes SEPN1-related myopathy. EMBO Rep. 2006;7:450-4.
- [CrossRef] [PubMed] [Google Scholar]
- Early onset myopathy with a novel mutation in the Selenoprotein N gene (SEPN1) Neuromuscul Disord. 2005;15:299-302.
- [CrossRef] [PubMed] [Google Scholar]
- The first report of two homozygous sequence variants in FKRP and SELENON genes associated with syndromic congenital muscular dystrophy in Iran: Further expansion of the clinical phenotypes. J Gene Med. 2020;22:e3265.
- [CrossRef] [PubMed] [Google Scholar]
- The phenotype and long-term follow-up in 11 patients with juvenile selenoprotein N1-related myopathy. Eur J Paediatr Neurol. 2008;12:224-30.
- [CrossRef] [PubMed] [Google Scholar]
- Scoliosis correction in an adolescent with a rigid spine syndrome: Case report. Spine (Phila Pa 1976). 2005;30:E623-8.
- [CrossRef] [PubMed] [Google Scholar]
- Early-onset myopathies: Clinical findings, prevalence of subgroups and diagnostic approach in a single neuromuscular referral center in Germany. J Neuromuscul Dis. 2017;4:315-25.
- [CrossRef] [PubMed] [Google Scholar]
- Floppy infant syndrome as a first manifestation of LMNA-related congenital muscular dystrophy. Eur J Paediatr Neurol. 2021;32:115-21.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J Med Genet. 2021;58:326-33.
- [CrossRef] [PubMed] [Google Scholar]
- Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology. 2012;78:1258-63.
- [CrossRef] [PubMed] [Google Scholar]
- Phenotype-Genotype analysis of Chinese patients with early-onset LMNA-related muscular dystrophy. PLoS One. 2015;10:e0129699.
- [CrossRef] [PubMed] [Google Scholar]
- Dropped head congenital muscular dystrophy caused by de novo mutations in LMNA. Brain Dev. 2017;39:361-4.
- [CrossRef] [PubMed] [Google Scholar]
- De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. 2008;64:177-86.
- [CrossRef] [PubMed] [Google Scholar]
- LMNA-associated myopathies: The Italian experience in a large cohort of patients. Neurology. 2014;83:1634-44.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital muscular dystrophy with dropped head phenotype and cognitive impairment due to a novel mutation in the LMNA gene. Neuromuscul Disord. 2014;24:529-32.
- [CrossRef] [PubMed] [Google Scholar]
- Distal acroosteolysis, poikiloderma and joint stiffness: A novel laminopathy? Eur J Hum Genet. 2016;24:1220-2.
- [CrossRef] [PubMed] [Google Scholar]
- Two children with “dropped head” syndrome due to lamin A/C mutations. Muscle Nerve. 2010;42:839-41.
- [CrossRef] [PubMed] [Google Scholar]
- New POMT2 mutations causing congenital muscular dystrophy: Identification of a founder mutation. Neurology. 2007;69:1254-60.
- [CrossRef] [PubMed] [Google Scholar]
- POMT1 and POMT2 mutations in CMD patients: A multicentric Italian study. Neuromuscul Disord. 2008;18:565-71.
- [CrossRef] [PubMed] [Google Scholar]
- The expanding phenotype of POMT1 mutations: From Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat. 2006;27:453-9.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12:2853-61.
- [CrossRef] [PubMed] [Google Scholar]
- Novel mutation in the fukutin gene in an Egyptian family with Fukuyama congenital muscular dystrophy and microcephaly. Gene. 2014;539:279-82.
- [CrossRef] [PubMed] [Google Scholar]
- A novel missense mutation in POMT1 modulates the severe congenital muscular dystrophy phenotype associated with POMT1 nonsense mutations. Neuromuscul Disord. 2014;24:312-20.
- [CrossRef] [PubMed] [Google Scholar]
- Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul Disord. 2009;19:182-8.
- [CrossRef] [PubMed] [Google Scholar]
- A Portuguese case of Fukuyama congenital muscular dystrophy caused by a multi-exonic duplication in the fukutin gene. Neuromuscul Disord. 2013;23:557-61.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical variation within sibships in Fukuyama-type congenital muscular dystrophy. Brain Dev. 1992;14:334-7.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic variations and clinical spectrum of dystroglycanopathy in a large cohort of Chinese patients. Clin Genet. 2021;99:384-95.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and orthopedic management of foot and ankle deformities in Charcot-Marie-Tooth disease. Muscle Nerve. 2018;57:255-9.
- [CrossRef] [PubMed] [Google Scholar]
- Facio-scapulohumeral muscular dystrophy with early joint contractures and rigid spine. Acta Myol. 2019;38:25-8.
- [Google Scholar]
- 12-Month progression of motor and functional outcomes in congenital myotonic dystrophy. Muscle Nerve. 2021;63:384-91.
- [CrossRef] [PubMed] [Google Scholar]
- A short form of gross motor function measure for Fukuyama congenital muscular dystrophy. Brain Dev. 2020;42:383-8.
- [CrossRef] [PubMed] [Google Scholar]
- Upper extremity outcome measures for collagen VI-related myopathy and LAMA2-related muscular dystrophy. Neuromuscul Disord. 2017;27:278-85.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile Pompe disease on ERT: Update on clinical presentation, musculoskeletal management, and exercise considerations. Am J Med Genet C Semin Med Genet. 2012;160C:69-79.
- [CrossRef] [PubMed] [Google Scholar]
- A consensus statement on the surgical treatment of Charcot-Marie-Tooth disease. Foot Ankle Int. 2020;41:870-80.
- [CrossRef] [PubMed] [Google Scholar]
- Comparison of long-term ambulatory function in patients with Duchenne muscular dystrophy treated with eteplirsen and matched natural history controls. J Neuromuscul Dis. 2021;8:469-79.
- [CrossRef] [PubMed] [Google Scholar]
- Orthopaedic disorders in myotonic dystrophy type 1: Descriptive clinical study of 21 patients. BMC Musculoskelet Disord. 2013;14:338.
- [CrossRef] [PubMed] [Google Scholar]
- Orthopedic management of patients with Pompe disease: A retrospective case series of 8 patients. ScientificWorldJournal. 2014;2014:963861.
- [CrossRef] [PubMed] [Google Scholar]
- Occurrence of Emery-Dreifuss muscular dystrophy in a rural setting of Cameroon: A case report and review of the literature. BMC Res Notes. 2017;10:36.
- [CrossRef] [PubMed] [Google Scholar]
- Linker protein repair of LAMA2 dystrophic neuromuscular basement membranes. Front Mol Neurosci. 2019;12:305.
- [CrossRef] [PubMed] [Google Scholar]
- Zebrafish models of LAMA2-related congenital muscular dystrophy (MDC1A) Front Mol Neurosci. 2020;13:122.
- [CrossRef] [PubMed] [Google Scholar]
- Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology-directed-repair-independent mechanism. Nat Med. 2017;23:984-9.
- [CrossRef] [PubMed] [Google Scholar]
- Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-α2 chain-null congenital muscular dystrophy mice. Hum Mol Genet. 2013;22:4914-28.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of candidate protein markers in skeletal muscle of laminin-211-deficient CMD type 1A-patients. Front Neurol. 2019;10:470.
- [CrossRef] [PubMed] [Google Scholar]
- Deletion of exon 4 in LAMA2 is the most frequent mutation in Chinese patients with laminin α2-related muscular dystrophy. Sci Rep. 2018;8:14989.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and genomic characteristics of LAMA2 related congenital muscular dystrophy in a patients' cohort from Qatar. A population specific founder variant. Neuromuscul Disord. 2020;30:457-71.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac involvement in Emery-Dreifuss muscular dystrophy and related management strategies. Int Heart J. 2019;60:12-8.
- [CrossRef] [PubMed] [Google Scholar]
- Dilated cardiomyopathy with conduction defects in a patient with partial merosin deficiency due to mutations in the laminin-α2-chain gene: A chance association or a novel phenotype? Muscle Nerve. 2011;44:826-8.
- [CrossRef] [PubMed] [Google Scholar]
- Management of respiratory complications and rehabilitation in individuals with muscular dystrophies: 1st Consensus Conference report from UILDM-Italian Muscular Dystrophy Association (Milan, January 25-26, 2019) Acta Myol. 2021;40:8-42.
- [Google Scholar]
- Rare variant in LAMA2 gene causing congenital muscular dystrophy in a Sudanese family. A case report. Acta Myol. 2019;38:21-4.
- [Google Scholar]
- Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin Genet. 2015;87:233-43.
- [CrossRef] [PubMed] [Google Scholar]
- Natural history, outcome measures and trial readiness in LAMA2-related muscular dystrophy and SELENON-related myopathy in children and adults: Protocol of the LAST STRONG study. BMC Neurol. 2021;21:313.
- [CrossRef] [PubMed] [Google Scholar]
- Muscle diseases with prominent joint contractures: Main entities and diagnostic strategy. Rev Neurol (Paris). 2013;169:546-63.
- [CrossRef] [PubMed] [Google Scholar]
- National registry of patients with Fukuyama congenital muscular dystrophy in Japan. Neuromuscul Disord. 2018;28:885-93.
- [CrossRef] [PubMed] [Google Scholar]




