Translate this page into:
Whole exome sequencing analysis identifies a missense variant in COL1A2 gene which causes osteogenesis imperfecta Type IV in a family from Saudi Arabia
2 Centre for Human Genetics, Hazara University, Mansehra, Pakistan
3 Department of Molecular Science and Technology, Ajou University, Suwon, South Korea
4 Department of Zoology, Islamia College University, Peshawar, Pakistan
5 Centre for Human Genetics, Hazara University, Mansehra; Department of Zoology, Islamia College University, Peshawar, Pakistan
6 Princess Al-Jawhara Albrahim Center of Excellence in Research of Hereditary Disorders, King Abdulaziz University, Jeddah, Saudi Arabia
Corresponding Author:
Musharraf Jelani
Princess Al-Jawhara Albrahim Center of Excellence in Research of Hereditary Disorders, King Abdulaziz University, Jeddah 21589
Saudi Arabia
mjelani@kau.edu.sa
| How to cite this article: Alkhiary YM, Ramzan A, Ilyas M, Khan U, Nasir A, Khan MI, Ahmad H, Jelani M. Whole exome sequencing analysis identifies a missense variant in COL1A2 gene which causes osteogenesis imperfecta Type IV in a family from Saudi Arabia. J Musculoskelet Surg Res 2017;1:33-38 |
Abstract
Objectives: Molecular diagnosis of a large Saudi family presenting an autosomal dominant form of osteogenesis imperfecta (OI). Methods: Genetic analysis of the index patient was performed through 100× paired end whole exome sequencing (WES) covering 24,000 coding genes of the human genome. The causative variant was filtered out among the previously known 23 genes' panel reported for 17 subtypes of OI. The dominant segregation of the causative variant with the disease phenotype was confirmed by Sanger sequencing. Pathogenicity of the altered protein was predicted through SIFT, PolyPhen, and MutationTaster software. Results: A heterozygous variant (c.1801G>A; p. Gly601Ser) in exon 31 of collagen 1α2 was identified. In this study, WES was successfully applied to identify the molecular basis of OI in the proband. The rest of family members were confirmed through Sanger validation confirming the autosomal dominant mode of inheritance in large Saudi family. Conclusion: OI is a rare heterogeneous disorder of connective tissues with 17 overlapping subtypes, for which 23 genes are known. Our work adds to the growing list of disease-causing variants in COL1A2. Reporting the disease-causing variants is one of the best ways to share data for better and accurate variants interpretation. We tested that WES can be used as an efficient tool for the molecular diagnosis of this rare phenotype.
Introduction
Osteogenesis imperfecta (OI) also known as brittle-bone disease, is a clinically and genetically heterogeneous group of connective-tissues' disorders. Its frequency is one out of 15,000‘20,000 live births.[1] OI mainly affects the bones, and of which symptoms may include bone fragility, increased susceptibility to bone fractures, growth deficiency, blue sclera, hearing deficit, dentinogenesis imperfecta (DI), skin and ligaments' hyperlaxity.[1] In 1978, Sillence first classified OI into four clinical and subclinical types based on the severity of bones involvement.[2] The severity of the condition is highly variable and ranges from mild-to-lethal phenotypes.[3] Type 1 is a nondeforming OI with blue sclera and is related to quantitative deficiency of normal collagen protein, whereas Type 2 is a lethal form, Type 3 is related to progressively deforming, and Type 4 is without blue sclera mainly result from mutations that alter the collagen structure.[1]
OI Type 4 (OI4, OMIM 166,220) is a generalized connective tissue disorder, and the patients are characterized by severe osteoporosis and bone fragility. Other features of the disease may include DI, scoliosis, short stature, hearing loss, skin, and ligament laxity; however, sclerae is not the feature of OI4.[4],[5] In 90% of OI cases, heterozygous variants of COL1A1 (OMIM 120,150) at chromosome 17q21.33 encoding collagen 1α1 and COL1A2 (OMIM 120,160) at chromosome 7q21.3 encoding collagen 1α2 have been reported causative for the phenotype.[3] Based on the structural or quantitative alterations in the collagen genes, the disorder is predominantly understood as a collagen-related disorder.[6],[7]
The laboratory investigations included collagen biochemical testing and mutation analysis of OI associated genes through next-generation sequencing (NGS). To date, mutations in 23 genes have been identified with OI and closely related disorders.[8] Whole exome sequencing (WES) analysis has been readily adopted as the first-line diagnostic tool in most of the Mendelian disorders worldwide.
In Saudi Arabia, as compared to autosomal dominant, the autosomal recessive cases are more prevalent due to high rates of consanguinity.[9] The WES analysis for various consanguineous families has been very recently utilized as a successful diagnostic tool.[10],[11],[12],[13],[14] In WES analysis, autosomal recessive disorders require the trio-analysis (father-mother-affected sibling), autosomal dominant disorders can be confirmed by analyzing patients in at least two or three successive generations.
In the current study, we ascertained a Saudi Arabian family segregating autosomal dominant phenotype of OI. Clinically, the disorder had been studied in Saudi Arabia who needed surgical corrections due to bone deformities;[15],[16] however, this is the first report to the best of our knowledge that has investigated WES analysis for the molecular diagnosis.
Material and Methods
Patients' enrollment
A five generations family segregating OI4 phenotype resided in Jeddah, Saudi Arabia [Figure - 1]. The patients had differentially been diagnosed with OI or DI. However, the clinical subtype of the disease was not clear. On obtaining the disease history from the family members, it was revealed that four generations had segregated the disease in autosomal dominant fashion. For genetic analysis, samples from nine family members (III-7, IV-7, IV-8, IV-11, IV-13, IV-14, V-1, V-2, and V-3) in which patients belonged to three generations, that is, III, IV and V were collected, and genomic DNA was extracted using standard methods.
![[Figure - 1]](#fig_SaudiOrthopJ_2017_1_2_33_218445_f1.jpg){kind=link}
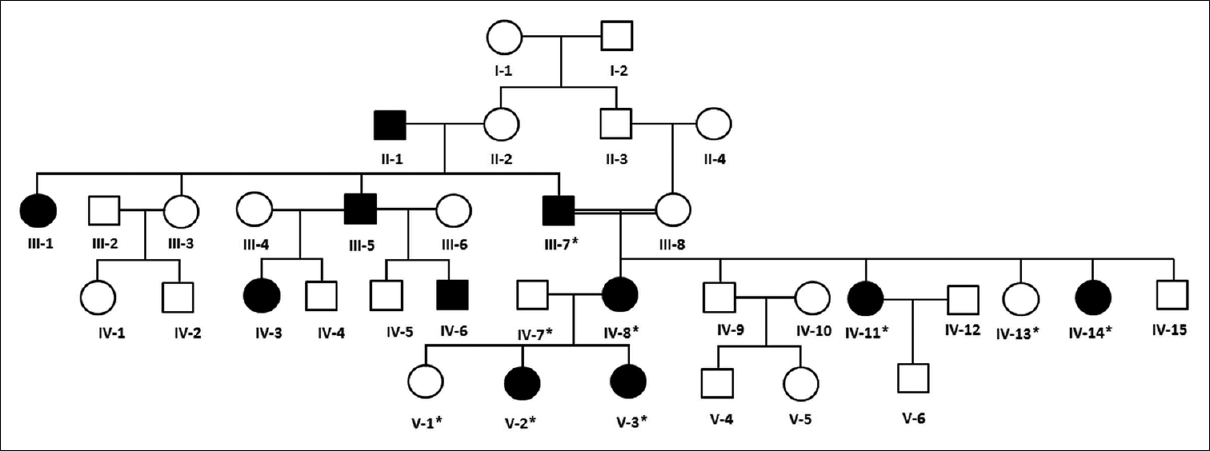 |
| Figure 1: Pedigree and haplotype analysis of the osteogenesis imperfecta family. Squares symbolize males and circles represent females. The filled symbols represent affected and the white are normal individuals. The individuals tested for COL1A2 mutations are highlighted with “*” in each case |
Clinical investigations
The affected individuals (the index patient IV-8 and her daughters V-2 and V-3) in the family were examined at the Oral and Maxillofacial Prosthodontics Department, Faculty of Dentistry and Department of Genetic Medicine, King Abdulaziz University for clinical diagnosis. The molecular genetics analysis for causative gene identification was performed at Princess Al-Jawhara Center of Excellence in Research of Hereditary Disorders, King Abdulaziz University, Jeddah, Saudi Arabia.
Molecular investigations
Whole exome sequencing and bioinformatics
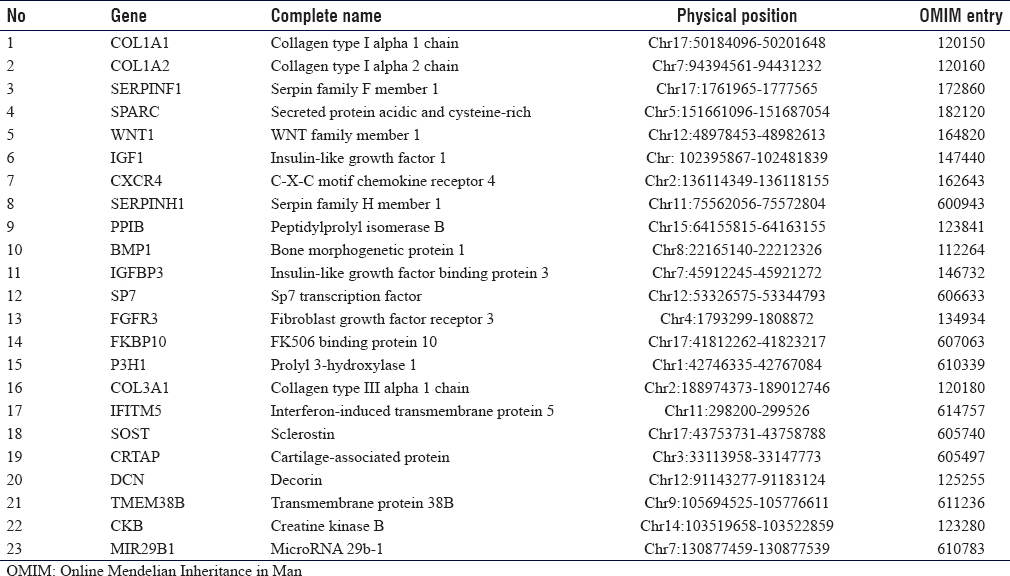
A total of 2 μg genomic DNA from the index patient was subjected to human whole exome analysis with paired-end sequencing at 100× resolution as described earlier.[12],[14] WES Libraries were created using 51 Mb SureSelect V6 kits (Agilent Technologies, Santa Clara, CA). Target regions with average throughput depths of more than 130 and 103 bp paired-end reads were sequenced using the HiSeq 2500 platform (Illumina, San Diego, CA, USA). For the alignment of sequences and copy number variations or the detection of small indels, standard software such as Burrows-Wheeler Aligner (http://bio-bwa.sourceforge.net/) and SAMTOOLS (http://samtools.sourceforge.net/) was used. The obtained reads were mapped to human UCSC genome database hg19 (http://genome.ucsc.edu/) and were equated with dbSNP (http://www.ncbi.nlm.nih. gov/snp/) and the 1000 Genomes (http://www. 1000 genomes.org/) databases. Filtering candidate variants in the coding regions, a dominant model was used to selected heterozygous alterations, the minor allele frequency was equal or < 0.0l, and predicted protein affect declared “damaging” were selected. The rest of the variants were filtered out. The predicted protein effect was also cross-checked with if involved in any of the OI disease phenotypes. For this reason, we first focused on the genes previously involved in OI and related phenotypes.[8] The list of these known genes included ALPL, ANO5, BMP1, CKB, COL1A1, COL1A2, COL3A1, CRTAP, CXCR4, DSPP, FGFR3, FKBP10, IFITM5, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SOST, SP7, TMEM38B, VDR, and WNT1.
Sanger sequencing for validation and population screening
Sanger sequencing was used to validate selected potentially causative variants in all available family members and ethnically matched healthy control chromosomes. Ensembl genome browser (http://www.ensembl.org/) was used to obtain the reference sequence of COL1A2 (ENST00000297268.10). The primer sequences (forward: 5'AGGGCTCGGAAGCTACAC-3' and reverse: 5'-GGCCAAACCAGCAATATAGA-3') for polymerase chain reaction amplification covering the candidate variant were designed using Primer3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/). The amplicon was amplified in each sample and screened by DNA cycle sequencing using a BigDye Terminator v3.1 Cycle Sequencing Kit in an ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequence variants were identified through BioEdit sequence alignment editor version 6.0.7 (http://www.mbio.ncsu.edu/bioedit/bioedit.html).
In silico three-dimensional protein modeling
The three-dimensional protein model for the wild and mutant collagen alpha-2(I) proteins (residues 540‘850) were developed using online software I-TASSER (http://zhanglab. ccmb. med. umich. edu/I-TASSER) and visualized by Discovery Studio 2016 software (version 2.0, Accelrys Software). The wild-type and mutant sequences were aligned through multiple-threading alignments tool Local Meta-Threading-server.
Results
The initial clinical diagnosis of the family was DI, which is one of the features of OI. On average, more than 90% of bases had phred score higher than 20, and there were 42,351 variants, which were filtered based on quality. The total number of reads was 5.4 Gb, and the mean depth was more than 100. The rest of the reads were not selected for further analysis. Furthermore, the filtering criteria mentioned above provided us with a heterozygous missense variant c.1801G>A in coding region (exon 31) of COL1A2 gene, which changed glycine to serine at 601 aa position [Figure - 2] and [Figure - 3]. This variant has been reported previously in OI patients.[17] This alteration was further confirmed for its correct segregation with the disease phenotype following the dominant mode of inheritance in three generations of the family [Figure - 1]. The variant was predicted as “disease-causing” or “protein-damaging” by in silico prediction software including SIFT, PolyPhen-2, MutationTaster and MutationAccessor. For population screening in our routine, we use a panel of 100 chromosomes (healthy unrelated individuals in Southwestern region of Saudi Arabia), to calculate the minor allele frequency for each novel variant in this population. However, we did not find this variant in control samples. The affected allele c.1801A was also not listed in the 1000 genome database (www.internationalgenome.org/) nor in the 60,706 unrelated individuals' exomes Exome Aggregation consortium database (http://exac.broadinstitute.org/).
![[Figure - 2]](#fig_SaudiOrthopJ_2017_1_2_33_218445_f2.jpg){kind=link}
![[Figure - 3]](#fig_SaudiOrthopJ_2017_1_2_33_218445_f3.jpg){kind=link}
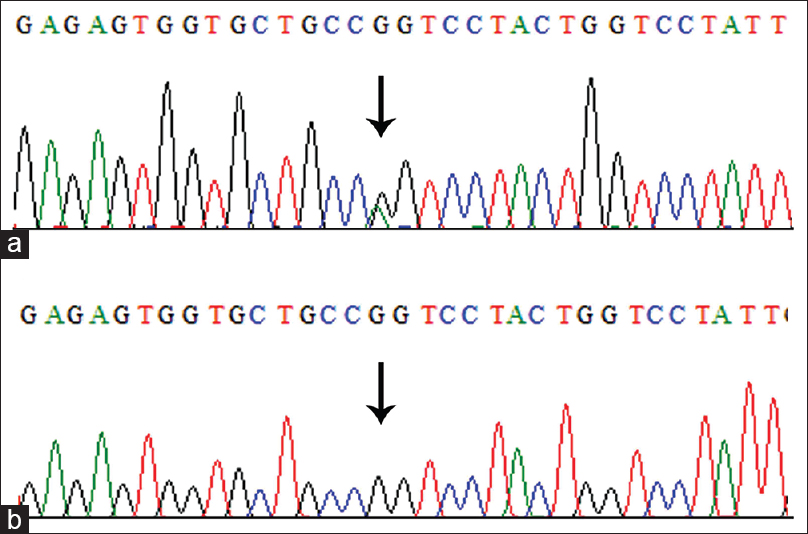 |
| Figure 2: Electropherogram of the missense mutation in COL1A2 gene (c.1801G>A) observed in patients with osteogenesis imperfecta (a) and wild type (b). The heterozygous nucleotide G>A substitution position 1801 is indicated by an arrow |
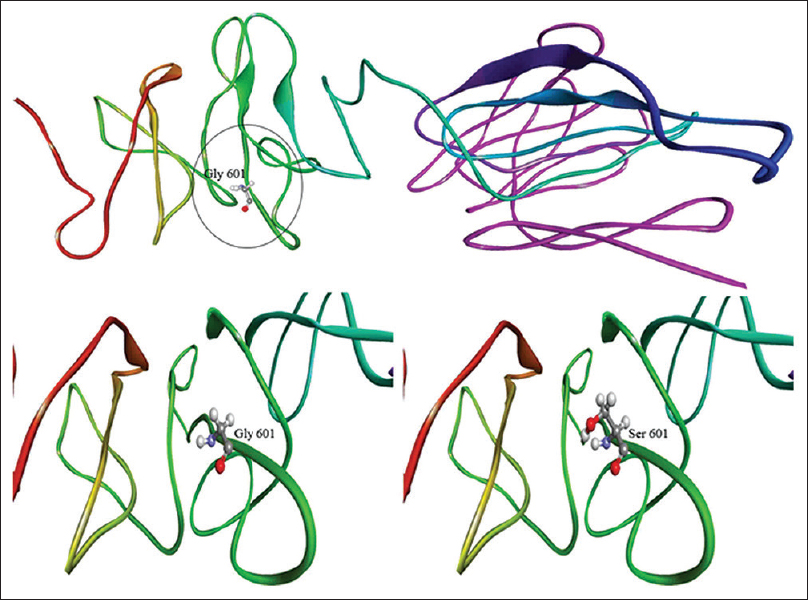 |
| Figure 3: Three-dimensional models of mutant and wild-type COL1A2 proteins. Note the encircles altered amino acid serine |
Discussion
In-time molecular diagnostics for various single gene disorders are becoming possible with the latest advances in NGS technologies. They are considered more efficient, accurate, and economical as compared to the earlier methods of candidate genes' analysis through Sanger sequencing. Rare Mendelian disorders are more frequent in isolated populations, and the future generations are at high risk of recurrence if proper genetic counseling or premarital testing is not available. In this study, through WES we tested a Saudi family for the causative gene identification leading to OI4 phenotype.
There are 17 clinical subtypes of OI for which 23 genes [Table - 1] are assigned so far. Based on the clinical overlap with OI and Sanger sequencing alone could lead to delays in obtaining results and were costlier. Thus, mutation screening of known genes through Sanger sequencing was skipped, and WES was performed as the first genetic test. The autosomal dominant model was used for candidate gene/variant prioritization. The variant we identified in COL1A2 (p. Gly601Ser) had been denoted as p. Gly511Ser in 2001 and was found in OI4 patients;[17] however, very recently a patient with OI1 has also been reported.[18] OI1 patients have blue sclera, which was not present in our family. The difference in the codon notation might be due to the difference in the older and newer versions of reference genome databases. The codon 601 has another variant c.1802G>A leading to glycine to aspartate alteration in OI4 patients.[19] In general, missense variants of COL1A1/A2 that changes glycine to serine have milder phenotype as reported in Estonian OI population.[20]
![[Table - 1]](#tbl_SaudiOrthopJ_2017_1_2_33_218445_t4.jpg){kind=link}
Apart from this, we also identified several alterations in dentin sialophosphoprotein (DSPP, OMIM 125,485) gene, which is previously known for DI.[21] Primarily, DI was one the differential diagnosis for these patients. This gave us an impression that DSPP might be the causative; however, these variants were also present in our in-house exome data of 24 healthy individuals. Thus, DSPP variants were declared neutral polymorphisms rather than potential candidates.
The COL1A2 encodes the α2-chain of type 1 collagen, which is a heterotrimeric protein consisting of two α1-chains (encoded by COL1A1) and one α2-chain. Type 1 collagen is the most abundant form of collagen in the human body and acts as the major structural protein of cartilage, bone, tendon, skin, and cornea.[22] Mutations in COL1A2 are the cause of a range of autosomal dominant conditions including OI2 and OI3 and autosomal recessive form of the cardiac-valvular type Ehlers‘Danlos syndrome.[23],[24],[25]
The precursor of type 1 collagen, called Type 1 procollagen consists of C- and N-terminal propeptides and a large central triple helix domain comprising a repeating [Gly-X-Y] triplet. The glycine residue in this triplet is the only amino acid small enough to reside within the sterically restricted inner aspect of the helix.[22] Most pathogenic missense mutations in COL1A1 and COL1A2 replace one of these crucial glycine residues with a larger amino acid, hence, leading to the synthesis of collagen with structural abnormalities. Alteration of a Gly part of a (Gly-X-Y) triplet typically causes more severe phenotypes as compared to mutations that lead to haploinsufficiency of Type 1 collagen (e.g., frameshift, nonsense, and splice-site mutations).[3],[26] The missense variant detected here in collagen alpha-2(I) chain causes a glycine to serine amino acid substitution at position 601 within the COL1A2 protein. The substitution of nonpolar Glycine (H side chain) by polar serine (OH side chain) can alter the intramolecular interactions of the protein structure. More specifically, this is an alteration of the glycine residue within a (Gly-X-Y) triplet comprising of amino acids Gly-Pro-Thr. Substitution of the small glycine in the larger serine is expected to affect the structure of the protein. This expected damaging effect is also reflected by the significant in silico predictions for this amino acid substitution. Apart from codon 601 glycine to serine changes at other positions of COL1A1/A2 had an adverse effect on collagen structure and function leading to teeth agenesis in OI.[18],[27],[28] An NGS molecular diagnostics panel, that is, 14 OI genes in Chinese cohort, revealed 73% of COL1A2 association in which most of the novel variants were an alternation of glycine residues.[29] This gives the importance of glycine amino acid in the collagen structure and function.
Conclusion
We emphasize that an in-time molecular diagnosis is a key to the future patient management and genetic counseling. Based on the genetic finding in the index case, at-risk family members were offered targeted genetic testing. When validated, WES analysis can be used in the near future as a first-tier molecular testing for OI4 and many other diseases with locus heterogeneity where single-nucleotide changes, small insertions or deletions are the predominant types of mutations.
Ethical approval and patients consent
The work was carried out in accordance with the Declaration of Helsinki, and ethical approval (project ref. 24‘14) was granted from the Medical Research and Ethics Unit, King Abdulaziz University (Jeddah, Saudi Arabia). Each participant above 18 years of age signed an informed written consent form to participate in the study and to publish clinical photographs and research for academic purposes. The legal guardian of participants below 18 years of age signed the consent letter.
Financial support and sponsorship
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant no.(4/165/1435-HiCi). The authors, therefore, acknowledge DSR for technical and financial support.
Conflicts of interest
There are no conflicts of interest.
Authors contributions
YMA enrolled the patients, made the clinical diagnosis and wrote the clinical synopsis. AR and MI contributed to the WES analysis. UK and AN contributed to the protein models' analysis. MIK and HA critically reviewed the manuscript. MJ designed the study, wrote and finalized the manuscript. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
| 1. | Forlino A, Marini JC. Osteogenesis imperfecta. Lancet 2016;387:1657-71. [Google Scholar] |
| 2. | Sillence DO, Rimoin DL. Classification of osteogenesis imperfect. Lancet 1978;1:1041-2. [Google Scholar] |
| 3. | van Dijk FS, Cobben JM, Kariminejad A, Maugeri A, Nikkels PG, van Rijn RR, et al. Osteogenesis imperfecta: A Review with clinical examples. Mol Syndromol 2011;2:1-20. [Google Scholar] |
| 4. | Cheung MS, Glorieux FH. Osteogenesis imperfecta: Update on presentation and management. Rev Endocr Metab Disord 2008;9:153-60. [Google Scholar] |
| 5. | Matzen K, Müller PK, Krieg T. Osteogenesis imperfecta: Biochemical characterization of various groups. Z Orthop Ihre Grenzgeb 1978;116:585-6. [Google Scholar] |
| 6. | Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 2011;7:540-57. [Google Scholar] |
| 7. | Marini JC, Blissett AR. New genes in bone development: What's new in osteogenesis imperfecta. J Clin Endocrinol Metab 2013;98:3095-103. [Google Scholar] |
| 8. | Van Dijk FS, Sillence DO. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014;164A: 1470-81. [Google Scholar] |
| 9. | Al-Owain M, Al-Zaidan H, Al-Hassnan Z. Map of autosomal recessive genetic disorders in Saudi Arabia: Concepts and future directions. Am J Med Genet A 2012;158A: 2629-40. [Google Scholar] |
| 10. | Alfares A, Alfadhel M, Wani T, Alsahli S, Alluhaydan I, Al Mutairi F, et al. A multicenter clinical exome study in unselected cohorts from a consanguineous population of Saudi Arabia demonstrated a high diagnostic yield. Mol Genet Metab 2017;121:91-5. [Google Scholar] |
| 11. | Ahmed S, Jelani M, Alrayes N, Mohamoud HS, Almramhi MM, Anshasi W, et al. Exome analysis identified a novel missense mutation in the CLPP gene in a consanguineous Saudi family expanding the clinical spectrum of Perrault Syndrome type-3. J Neurol Sci 2015;353:149-54. [Google Scholar] |
| 12. | Alkhiary YM, Jelani M, Almramhi MM, Mohamoud HS, Al-Rehaili R, Al-Zahrani HS, et al. Whole-exome sequencing reveals a recurrent mutation in the cathepsin C gene that causes Papillon-Lefevre syndrome in a Saudi family. Saudi J Biol Sci 2016;23:571-6. [Google Scholar] |
| 13. | Jelani M, Kang C, Mohamoud HS, Al-Rehaili R, Almramhi MM, Serafi R, et al. A novel homozygous PTH1R variant identified through whole-exome sequencing further expands the clinical spectrum of primary failure of tooth eruption in a consanguineous saudi family. Arch Oral Biol 2016;67:28-33. [Google Scholar] |
| 14. | Serafi R, Jelani M, Almramhi MM, Mohamoud HS, Ahmed S, Alkhiary YM, et al. Identification of two homozygous sequence variants in the COL7A1 gene underlying dystrophic epidermolysis bullosa by whole-exome analysis in a consanguineous family. Ann Hum Genet 2015;79:350-6. [Google Scholar] |
| 15. | Khoshhal KI, Ellis RD. Functional outcome of sofield procedure in the upper limb in osteogenesis imperfecta. J Pediatr Orthop 2001;21:236-7. [Google Scholar] |
| 16. | Khoshhal KI, Ellis RD. Effect of lower limb sofield procedure on ambulation in osteogenesis imperfecta. J Pediatr Orthop 2001;21:233-5. [Google Scholar] |
| 17. | Ward LM, Lalic L, Roughley PJ, Glorieux FH. Thirty-three novel COL1A1 and COL1A2 mutations in patients with osteogenesis imperfecta types I-IV. Hum Mutat 2001;17:434. [Google Scholar] |
| 18. | Andersson K, Dahllöf G, Lindahl K, Kindmark A, Grigelioniene G, Šström E, et al. Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta - A retrospective cohort study. PLoS One 2017;12:e0176466. [Google Scholar] |
| 19. | Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 2007;28:209-21. [Google Scholar] |
| 20. | Zhytnik L, Maasalu K, Reimann E, Prans E, Kõks S, Märtson A, et al. Mutational analysis of COL1A1 and COL1A2 genes among Estonian osteogenesis imperfecta patients. Hum Genomics 2017;11:19. [Google Scholar] |
| 21. | Takagi Y, Sasaki S. Histological distribution of phosphophoryn in normal and pathological human dentins. J Oral Pathol 1986;15:463-7. [Google Scholar] |
| 22. | Malfait F, Symoens S, Goemans N, Gyftodimou Y, Holmberg E, López-González V, et al. Helical mutations in type I collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos Syndrome overlap syndrome. Orphanet J Rare Dis 2013;8:78. [Google Scholar] |
| 23. | Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A, et al. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. J Med Genet 2006;43:e36. [Google Scholar] |
| 24. | Faqeih E, Roughley P, Glorieux FH, Rauch F. Osteogenesis imperfecta type III with intracranial hemorrhage and brachydactyly associated with mutations in exon 49 of COL1A2. Am J Med Genet A 2009;149A: 461-5. [Google Scholar] |
| 25. | Forlino A, Keene DR, Schmidt K, Marini JC. An alpha2(I) glycine to aspartate substitution is responsible for the presence of a kink in type I collagen in a lethal case of osteogenesis imperfecta. Matrix Biol 1998;17:575-84. [Google Scholar] |
| 26. | Rauch F, Lalic L, Roughley P, Glorieux FH. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res 2010;25:1367-74. [Google Scholar] |
| 27. | Lindahl K, Šström E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren Ö, et al. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet 2015;23:1042-50. [Google Scholar] |
| 28. | Malmgren B, Andersson K, Lindahl K, Kindmark A, Grigelioniene G, Zachariadis V, et al. Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes. Oral Dis 2017;23:42-9. [Google Scholar] |
| 29. | Liu Y, Asan, Ma D, Lv F, Xu X, Wang J, et al. Gene mutation spectrum and genotype-phenotype correlation in a cohort of chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int 2017;28:2985-95. [Google Scholar] |
Fulltext Views
4,077
PDF downloads
512




